Synthesis of Drugs¶
Info
By creating new molecules and synthesizing new drug candidates, chemistry plays a central role in drug discovery and development. This chapter introduces the strategies and tactics used in organic synthesis. Concepts such as retrosynthesis, reaction classifications, protecting groups, racemic or asymmetric syntheses and directed organic syntheses are described and illustrated with examples. The syntheses of some common drugs are also presented. The chapter reviews some of the computer programs available in automated synthesis planning and current databases used in organic synthesis.
Number of Pages: 198 (±4 hours read)
Last Modified: September 2005
Prerequisites: None
Introduction¶
Why to Synthesize a New Molecule?¶
Synthetic organic chemistry is a powerful field that governs much of drug discovery and development. It enables us access to new chemical compounds such as analogs of natural products or novel synthetic structures that can be used as probes to understand biological events and the derivation of useful therapeutic agents. It also plays a pivotal role in the preparation of the necessary quantities for clinical trials and bulk production of the drug when it becomes commercialized.

book
Douglas S. Johnson and Jie Jack Li
John Wiley and Sons, Hoboken 2007
Drug Discovery¶
The synthesis of new molecules plays a key role in the drug discovery process. A drug candidate results from a lengthy and intense research activity where many molecules are synthesized and extensively tested. This generates informative structure-activity relationships for the conversion of this knowledge in the optimization and development of the drug candidate.

Bulk Production¶
The manufacturing and bulk production of a drug is costly and involves heavy industrial installations. Millions of dollars can be saved by reducing the production costs of a drug. For example, for more than 60 years the commercial synthesis of Vitamin C has involved a six-step chemical process. Recently, an intelligent modification of the synthetic process has shortened it to only three major steps. More than 110,000 tons of Vitamin C are produced annually; the economical and ecological impact on what has become a $1 billion-plus industry are considerable.

Goal of the Synthesis¶
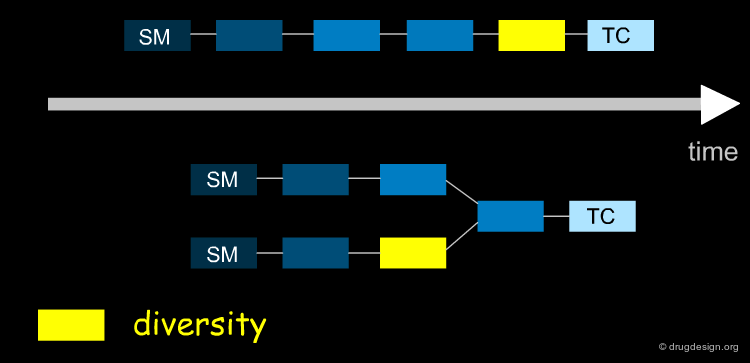
The aim of the synthesis is to prepare a target compound (TC, the drug candidate) which is synthesized by converting the starting material (SM) after a series of chemical interconversions have been carried out. A, B ... are intermediate products, whereas X, Ψ, Ω ... represent the reagents and conditions of the chemical reactions.

General Requirements before Starting¶
Before starting an organic drug synthesis, the requirements are generally well established. The target compound should be of low molecular weight. We need cheap, non toxic, readily available materials and keep the synthesis short. Whenever possible, stereospecific reactions with high yields must be planned. The reactions should be adaptable to large-scale production methods with intermediate products that are easy to isolate, purify and identify.

Number of Steps and Intermediates¶
Drug synthesis is a multiple step process. The strategy consists of moving from SM to TC with a minimum number of steps. It is extremely important to address the issue of reducing the number of steps: in drug discovery this accelerates the discovery cycle, and in process manufacturing this reduces production costs of the drug. For example, nifedipine is synthesized in 2 steps, viagra and zyvox in 9, lipitor and cephalexin in more than 11 steps.

Measurable Reaction's Characteristics¶
Many characteristics of a reaction can be observed and measured as the reaction proceeds. These typically include the yield, the reaction rate, the intermediates formed and the products obtained. Some of these are discussed in the following pages.


Yield¶
The yield of a reaction is a percentage that indicates the amount of product actually obtained as compared to the theoretical yield (which is the amount of product predicted to form on the basis of the balanced chemical equation). A yield of 80% is good per step. Note that a five step reaction with an average yield of 80% for each step would give an overall yield of only 33%: this shows that overall yields drop off rapidly even with high yields at each stage. It may be possible to increase the effectiveness of a given step by changing the experimental conditions such as the reagents, the temperature, the time or the catalysts.

Reaction Rate¶
Chemical reactions occur at different rates. Some reactions are extremely fast while others are very slow, depending on the nature of the reactants and products and the conditions under which the reaction takes place. Factors that affect reaction rates include temperature, the concentration of the reactants, the pH, the physical state of reactants, the presence of a catalyst and light.
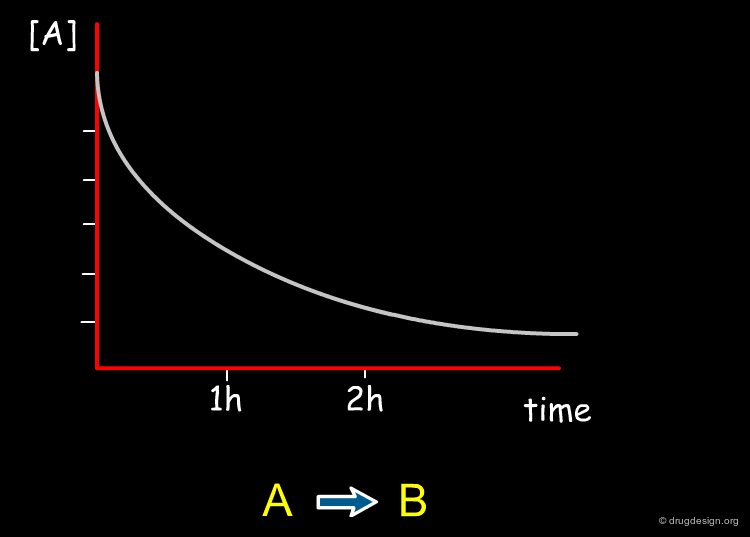
Product Selectivity¶
The result of a reaction between a reactant and a reagent is not always a unique product; multiple competing reactions can take place and produce different products. Normally for the synthesis of a drug, only one of the products is needed and the reaction is optimized to maximize the formation of the desired compound as far as possible.

Regioselectivity and Regiospecificity¶
Above we saw that a reaction can yield different products, and these products are not isomers. However, there are reactions that produce isomeric products, such as regioselective reactions. A simple example is illustrated below, where the reaction of HBr with propene gives 1-bromopropane and 2-bromopropane (if this regioselective reaction had given only one product it would have been called "regiospecific").
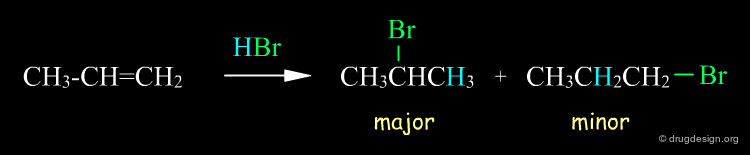
Stereoselectivity and Stereospecificity¶
A reaction is stereoselective when there is preferential formation of one stereoisomer over another. A reaction is sometimes termed stereospecific when there is very high stereoselectivity. Stereoselectivity is an important variable in the synthesis of many drugs.

Thermodynamic and Kinetic Properties of the Reaction¶
Thermodynamics and kinetics control the course of every chemical reaction. A reaction can be thermodynamically controlled (when the product composition is governed by the equilibrium thermodynamics of the system), or kinetically controlled (when the product composition is governed by competing rates of formation of products), in which case the formation of the product with the lowest activation energy is favored.

Thermodynamics¶
The enolization of keto-3 steroids is an example of a thermodynamically controlled reaction. The composition of the two enols obtained is dictated by their relative energies: there is a difference of 2.4 kJ/mol, in favor of the Δ2 enol.

articles
249. Steroids. Part CLXXI. Factors Controlling the Direction of Enol Acetylation of 3-oxo-steroids Berkoz B, Chavez EP and Djerassi C J. Chem. Soc.
1962
The Torsion Angle Concept in Conformational Analysis Bucourt R Topics in Stereochemistry 8 1974
Kinetics¶
The nucleophilic attack of a ketone in a chiral environment is under kinetic control: unequal amounts of enantiomeric alcohols are produced because the two diastereoisomeric transition states do not have the same energy.

Determinants of a Chemical Reaction¶
Thermodynamic and kinetic properties determine the course of a chemical reaction. The reaction is driven by the energies of the reagents and products (thermodynamics) and of the transition states (kinetics). Steric, electronic and solvent effects for instance contribute to the energies of such molecular systems.

Steric Effects¶
Steric effects can influence conformational preferences or stabilizing transition states of reactions. They control the course of the regioselectivity of many reactions such as additions, eliminations, substitutions or rearrangements. For example the regioselective formation of the less crowded alkene in the following Hofmann elimination is due to steric effects resulting in the low energy of the corresponding transition state.

Electronics Effects¶
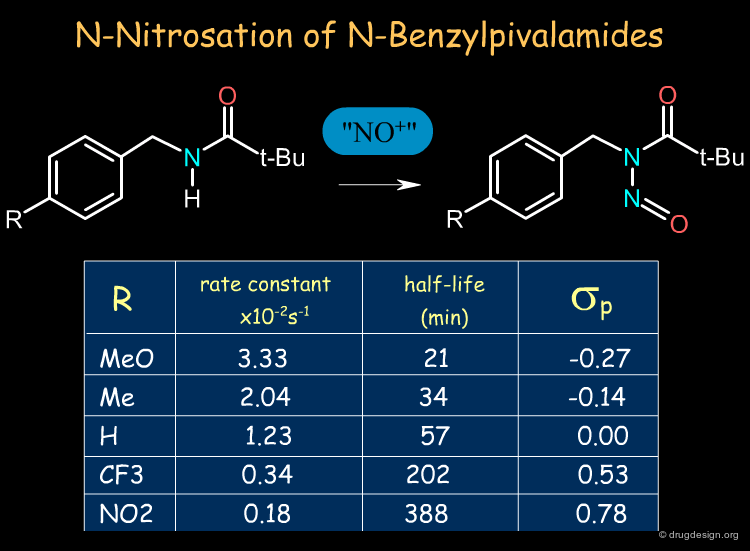
Electronic effects can influence the resonance, the inductive effects, the stereoelectronic features, the basicity of nucleophiles, the acidity of electrophiles, the stabilization of ionic structures etc... The classical case is the ortho, meta or para substitutions in Friedel Crafts reactions. In the example below we see how the rate of N-Nitrosation of N-Benzylpivalamides can be accelerated by para electron-donating groups.
articles
Electronic Effects in the N-Nitrosation of N-Benzylpivalamides Ron W. Darbeau, Rebecca S. Pease, and Edson V. Perez J. Org. Chem. 67 2002
Solvent Effects¶
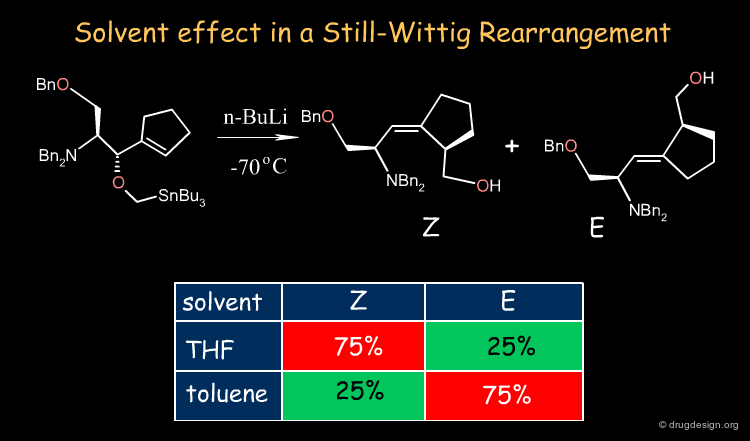
The solvent performs the mechanical but vital role of allowing immiscible reactants to come together rapidly. The example shown below illustrates the influence of the solvent in a Still-Wittig rearrangement yielding opposite stereoselectivities depending on the nature of the solvent.
articles
Solvent-Dependent Stereoselectivity in a Still-Wittig Rearrangement: An Experimental and ab Initio Study Scott A. Hart, Carl O. Trindle, and Felicia A. Etzkorn Organic Letters 3 2001
How to Influence a Reaction?¶
Because of their complexity, the chemist is less concerned with the energies of the products or the transition states, and is more interested in practical ways of influencing the reaction so it can be oriented towards the desired end products. The concrete parameters that the chemist can influence are for example the choice of the reactants, reagents, concentrations, temperature, catalyst, pH, solvent, etc...

Reactant Choice¶
Once a reaction has been selected it can be influenced by changing various experimental parameters - first and foremost, the choice of the reactants and reagents. In the example below (molecules that are important in the discovery of EGF-R kinase inhibitors) the nature of the halogen in position 6 proved to be critical for enabling the reaction to take place. A fluorine atom in position 6 is easily displaced by primary and secondary amines, whereas chlorine in the same position cannot be displaced by amines or other nucleophiles.
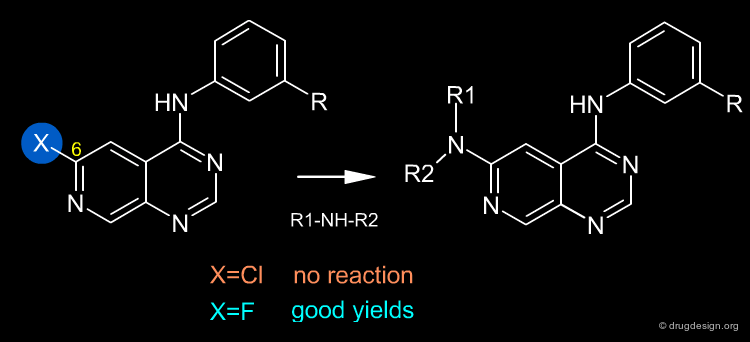
articles
Synthesis of 4-(Phenylamino)pyrimidine Derivatives as ATP-Competitive Protein Kinase Inhibitors with Potential for Cancer Chemotherapy Rewcastle G.W.; Denny W.A.; Showalter H.D.H. Current Organic Chemistry 4 July 2000
Reagent Choice¶
The example below illustrates how it is possible to direct the stereoselectivity of a reaction by modifying the reagent. It shows how it is possible to influence the course of the reaction towards the desired product with the right choice of the nucleophile.

Reaction Conditions¶
Once a specific reaction has been selected, it is implemented - which is one of the chemist's most time consuming jobs. A whole set of conditions need to be defined: the reagents, the time, the temperature, the concentration or the physical state of the reactants, the solvent, the pH, the catalysts and their combinations to obtain the desired product. In the example below, 50 trials were carried out before the best set of conditions (with a yield of 96%) was found. In unfavorable conditions, the reaction does not work well and cannot be rectified.

Influence of pH¶
The following example illustrates how a Friedel-Crafts acylation in water can be sensitive to pH. At pH 7.5 the indolic system is efficiently acylated with ethyl glyoxylate. At this pH the yields are as high as 95%; they drop to 17% at pH 11.3.

articles
Friedel-Crafts reactions in water of carbonyl compounds with heteroaromatic compounds Wei Zhuang and Karl Anker Joergensen Chemical Communications 13 2002
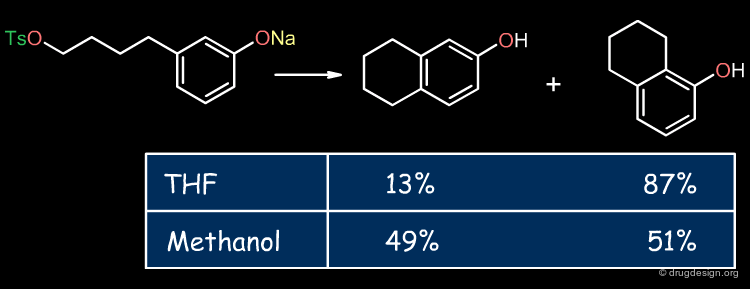
Influence of the Solvent¶
The solvent can substantially affect the solvation of reactants, products or transition states resulting in variations in the regioselectivity, reaction rate or equilibrium of a reaction. From solvent to solvent the solvation energies may vary by up to 40 kJ/mol.


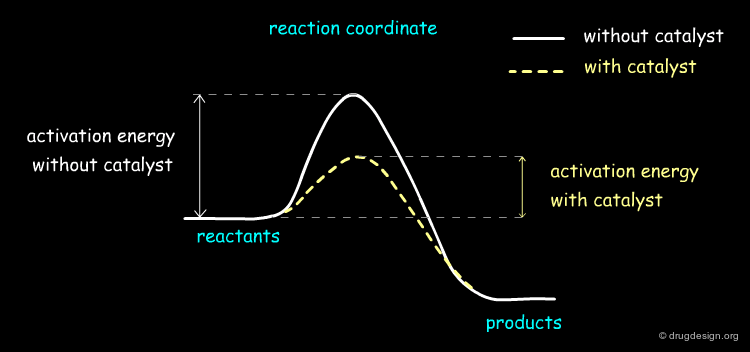
Catalysts¶
The role of the catalyst is to facilitate the formation of the desired product by reducing the activation energy of the reaction. Over the last few decades chemists have devised a vast range of chiral and non-chiral catalysts that contain complex inorganic, organic and biological catalysts that are designed for specific reactions such as Suzuki coupling, asymmetric metathesis etc....


Tools for Following the Progression of a Reaction¶
Because a reaction takes place in many steps, it is important to have the right tools to make sure that the compounds obtained at each step are the expected ones. Did the reaction take place? Was the reaction complete? How many products are in the flask? What is the main product? These are some of the questions which need to be answered. There is a broad range of analytical tools available to monitor the progression of a chemical reaction. Spectroscopy and chromatography are the most indispensable means used in all laboratories. It is possible to rely on a single analytical method or in a combination or several of them.

Spectroscopy¶
Spectroscopy is an important tool to test for the correct progression of the synthesis of a drug. A spectrum can be obtained (absorption or emission), that provides a fingerprint that is characteristic of the molecular structure concerned. It can be compared to the spectrum of a known compound, or analyzed in terms of the chemical structure of the compound considered. Analytical spectroscopic tools include mass spectroscopy, infrared, nuclear magnetic resonance, ultraviolet, circular dichroism and X-ray crystallography.

book
D. Williams and I. Fleming
McGraw-Hill 1989
Mass Spectra (MS)¶
In mass spectrometry, a substance is bombarded with an electron beam with sufficient energy to fragment the molecule. The ions generated are accelerated through a magnetic field and are sorted on the basis of a mass-to-charge ratio. A precise molecular mass is obtained and the analysis of the spectrum involves re-assembling the fragments to generate the exact structure of the original molecule.

Infrared (IR)¶
Absorption of infrared radiation results in the excitation of vibrational, rotational and bending modes of groups of atoms, while the molecule itself remains in its electronic ground state. The functional groups present in the molecule can easily be identified through their characteristic absorption wavelengths.

Nuclear Magnetic Resonance (NMR)¶
Nuclear magnetic resonance is a spectroscopic technique that provides information about the number and types of atoms in a molecule. NMR spectrometers are tuned to certain nuclei such as 1H, 13C, 19F and 31P. Absorption in the low-energy radio-frequency part of the spectrum causes excitation of nuclear spin states; high-resolution spectroscopy distinguishes atoms in different locations in the molecule.

Ultraviolet (UV)¶
Absorption or emission of ultraviolet or visible light by a molecule depends on electronic transitions between molecular orbital energy levels in the molecule. UV spectra can be used to identify an unknown compound by comparative analyses. It is not used to identify the presence of functional groups, but rather to show relationships as conjugation. UV spectra may be also used to determine the concentration of a compound in a mixture.

Circular Dichroism (CD) and Optical Rotatory Dispersion (ORD)¶
Circular dichroism is used for the spectroscopic analysis of chiral molecules by measuring differences in the absorption of left-handed polarized light versus right-handed polarized light, which arise from the structural asymmetry of the chiral moieties.

X-rays¶
Solid state X-ray crystallography is used to reveal the structure of a compound, information that could be invaluable when its 2D formula is problematic. For example, Dorothy Hodgkin and her team at Oxford were able to demonstrate the structure of penicillin by the resolution of its X-ray diffraction data. Dorothy Hodgkin was awarded the Nobel Prize in chemistry (in 1964) for this achievement that enabled chemists to develop synthetic ways to make this antibiotic - and many related analogs.

Chromatography¶
Analytical chromatography is essential for following the progression of a synthesis. Based on differential migration of solutes, it can identify reagents, reactants and products that are present in the reaction mixture (by comparing it with the corresponding pure compounds). The technique was developed in the 1940s with paper chromatography, and in the 1950s with gas (GC) and thin layer chromatography (TLC). Today modern chromatography has generated a broad spectrum of techniques including high performance liquid chromatography (HPLC) for both analytical and preparative purposes.

Media
Actelion Pharmaceuticals Ltd., Reproduced from Actelion, with permission Actelion
High Performance Liquid Chromatography (HPLC)¶
High performance liquid chromatography is a tool for quantifying and analyzing mixtures of chemical compounds. The method was developed in the mid-1970s and has become the most popular method of analysis because it is easy to use.

Chiral Chromatography¶
The separation of enantiomers by chiral HPLC is a useful method for the analysis of chiral substances. It can be a very efficient method for the separation of racemic compounds, to control the optical purity and also for the preparation of optically pure drugs.

Design Strategy¶
Strategy Like a General in the Battle¶
The synthesis of the target compound can be viewed as a battle in which the strategy that is best suited to achieving the upcoming goal is applied. The current status must be permanently assessed and the strategy revised. The medicinal chemist must find the appropriate reagents at minimum price and the best synthetic scheme to achieve this goal as quickly as possible.

Flexibility in the Strategy¶
Many syntheses contain surprises and the chemist must be prepared to circumvent difficulties. "Flexibility" is the keyword and must be present at multiple levels, as discussed in the following pages.

Flexibility in the Synthetic Program¶
In terms of chemistry, the synthetic scheme must be as flexible as possible. As many problems as possible must be predicted in advance at the design stages of a chemical program, so that there are alternative solutions available if a reaction fails to work.

Flexibility in the Target Molecule¶
Flexibility should be uppermost in the chemist's mind. After all a chemist "synthesizes" an idea rather than a molecule... In chemistry if the result diverges from the expected outcome, you need to return to the hypothesis. Based on accumulated knowledge and imagination, another molecule must be designed that fits the working hypotheses better than the original one.

Nicolaou Statement on the Trip to Ithaca¶
K.C. Nicolaou use to say says that organic synthesis is like the journey from Troy to Ithaca: this trip is full of adventure and new discoveries; encounters with Laistrygonians and Cyclops... For the medicinal chemist a second journey begins when the molecule enters the cycle of biological testing...

Linear and Convergent Strategy¶
The synthesis of the molecule is like putting together a jigsaw puzzle. You can assemble complementary blocks, or you can make certain regions independently and then connect them. In organic synthesis, these two approaches are known as the linear and convergent strategies.

Linear Strategy¶
In a linear strategy the molecule is constructed in a stepwise fashion, following a linear reaction sequence. The global yield of a linear synthesis decreases exponentially with the number of steps. Brevetoxine can be synthesized in 83 steps with an average yield of 91% per step, however the global yield is only 0.043% !

Example: Captopril Linear Synthesis¶
Captopril can be synthesized in four steps according to the linear synthesis shown below, that starts from 2-methylacrylic acid.

articles
Design of specific inhibitors of angiotensin converting enzyme: new class of orally active antihypertensive agents. Ondetti MA, Rubin B and Cushman DV. Science 196 1977
Convergent Strategy¶
In the convergent synthesis two or more direct precursors of a target are constructed independently and then assembled in the last step of the synthesis.

Convergent Advantage¶
Convergent routes present advantages over linear approaches with respect to speed, time, yields and manpower. Furthermore they have greater flexibility for producing series of analogs. The impact of low yield reactions is less dramatic in a convergent than in a linear approach, as illustrated in the theoretical examples below.

Example: Losartan Convergent Synthesis¶
Losartan can be synthesized in a convergent manner as shown below. The imidazole and biphenyl precursors 4 and 8 are first prepared separately from (1 and 2) and (5 and 6), respectively, and they react together in one of the last steps of the synthesis to give the cyano analog 9, which is then transformed into the desired losartan drug 10 with the desired tetrazole moiety.



articles
Nonpeptide angiotensin II receptor antagonists: the discovery of a series of N-(biphenylylmethyl)imidazoles as potent, orally active antihypertensives David J. Carini, John V. Duncia, Paul E. Aldrich, Andrew T. Chiu, Alexander L. Johnson, Michael E. Pierce, William A. Price, Joseph B. Santella, III, Gregory J. Wells, and et al. J.Med.Chem.
1991
Nonpeptide Angiotensin II Receptor Antagonists: The Next Generation in Antihypertensive Therapy Ruth R. Wexler, William J. Greenlee, John D. Irvin, Michael R. Goldberg, Kristine Prendergast, Ronald D. Smith, and Pieter B. M. W. M. Timmermans J.Med.Chem. 39 1996
How to Analyze a Molecule for Synthesis?¶
Molecules have their own identity and capabilities and the chemist assesses the molecular complexity of the target in terms of characteristics such as those indicated in the list below. However, the analyses depend entirely on the chemist's experience and creativity in combining chemical reactions and selecting the best of a set of possible routes to making the molecule.
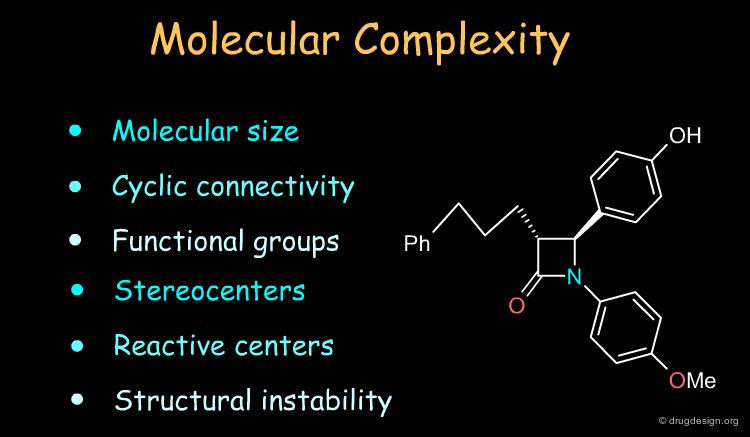
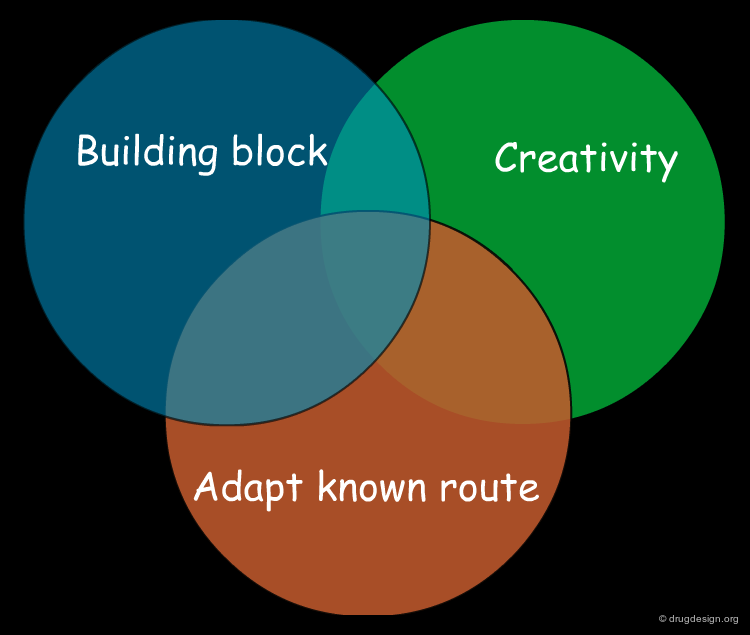
Three Methods for the Design of a Synthetic Program¶
Having assessed the complexity of the target molecule, the next step is to find how to synthesize it. There are three basic methods: (1) adapt a known synthetic scheme; (2) use a building block strategy and (3) start from scratch.

Adapt Known Synthetic Schemes¶
Adapting known synthetic schemes involves using available synthetical knowledge about other molecules. Rather than re-inventing the wheel, the chemist looks for similar molecules that have already been synthesized by other groups and decides whether the existing methods can be adapted for the synthesis of the target compound.

Literature and Patent Searches¶
Chemical Abstracts, Beilstein, literature and patent searches provide useful ways for finding synthetic schemes that could be adapted for the preparation of the new molecule.

Database Searching with Computer Programs¶
Today most information can be accessed through specialized databases. Database searching provides effective means for finding synthetic pathways and is a source of inspiration for the preparation of target compounds.

Consider a Building Block Strategy¶
Unlike the existing knowledge adaptation of known synthetic schemes, the building block strategy aims at exploiting existing starting material. The building block strategy consists of incorporating elaborated and readily available fragments in the synthesis. The recognition of useful building blocks for a target molecule is of outmost importance because it can substantially simplify the chemical pathway.

Small Commercial Building Blocks¶
The small building blocks for the synthesis of a novel drug must be commercially available and cheap. This is an issue that a medicinal chemist must constantly keep in mind.

Elaborated Building Blocks¶
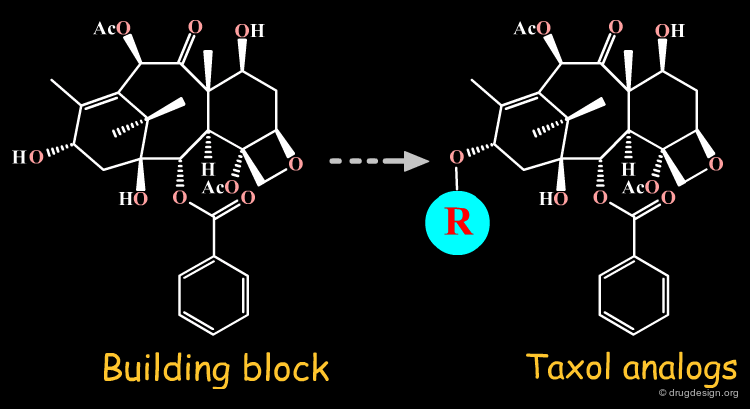
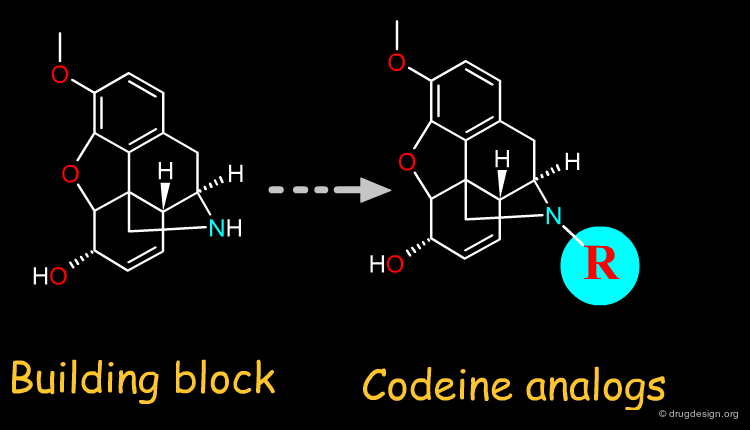
For semi-synthesis purposes, elaborated building blocks can be obtained from naturally occurring compounds. A great number of analogs can then be prepared with such scaffolds. Examples of moieties that can be either purchased or prepared are shown here in the taxol, codeine, staurosporine and penicillin families.


Starting from Scratch - Retrosynthetic Analysis¶
When no analogy with a known molecule and no building block can be found, a synthetic route has to be invented 'from scratch'. Although the design is relatively straightforward for a simple structure, a method is needed when the molecule is complicated. "Retrosynthetic analysis" is an important method for analyzing and partitioning the problem. This is presented in the following pages.

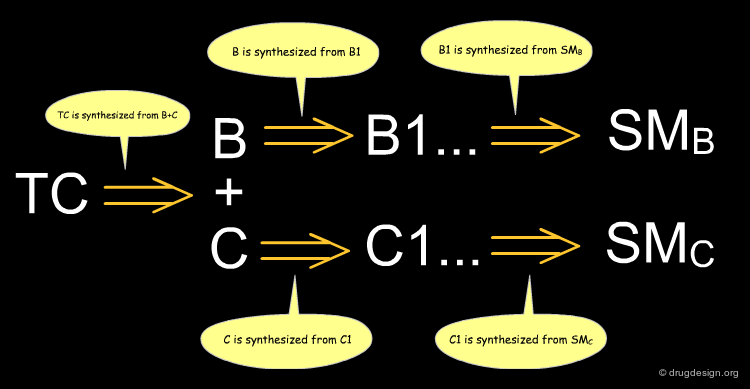
Retrosynthetic Strategy¶
In retrosynthetic analysis, a synthesis is planned by reasoning backward from the desired product. The approach was formalized by E.J. Corey (1990 Nobel Prize in Chemistry) by using standard rules. The steps are described using retrosynthetic arrows drawn as => and meaning "is synthesized from".

articles
Computer assisted analysis of complex synthetic problems E. J. Corey Q. Rev. Chem. Soc. 25 1971
Towards the ideal synthesis P. A. Wender, S. T. Handy and D. L. Wright Chem. Ind. (London)
1997
he Art and Science of Organic Natural Products Synthesis K. C. Nicolaou, E. J. Sorensen and N. Winssinger J. Chem. Educ. 75 1998
The Art and Science of Total Synthesis at the Dawn of the 21st Century K. C. Nicolaou, D. Vourloumis, N. Winssinger and P. S. Baran Angew. Chem. Int. Ed. 39 2000
book
E. J. Corey and X.-M. Cheng
Wiley, New York 1995
K. C. Nicolaou, Eric J. Sorensen
Wiley-VCH 1996
K. C. Nicolaou, Scott A. Snyder
Wiley-VCH 2003
Tse-Lok Ho
Wiley 1994
Stuart Warren
John Wiley and Sons 1978
Stuart Warren
Cambridge Univ. 1982
Elsevier Vol 1: 1984; Vol.5:2004
Disconnection of Strategic Bonds¶
In retrosynthesis the bonds of the target molecule are systematically disconnected to reveal the structures of precursor molecules (in orange in the figure), which are then examined in a similar way. One particularly useful tool in identifying good disconnections is to look at the relationship between functional groups. The key bonds that reveal the synthetic strategy are known as the "strategic bonds" and the reactive components of a synthetic step are called "synthons".

Strategic Bonds Revealed by Small Modifications¶
The aim of retrosynthesis is to find disconnections and uncover strategic bonds that lead to simple starting material. Sometimes a disconnection can only be found if a small modification of a functional group is introduced into the molecule (in the example below, A was changed to A'). The most common transformations are: functional group addition (FGA), functional group interconversion (FGI) and functional group removal (FGR).

FGA¶
An example of functional group addition (FGA) is illustrated below. The transform introduced (addition of a methyl ester next to the carbonyl) cleverly increases the complexity of the target molecule: a Dieckmann reaction on the cyclobutane diester derivative leads to the desired structure.

FGI¶
An example with successive functional group interconversions (or interchanges) (FGI) is shown below. Finally, the target compound can be prepared from acetone as starting material.

FGR¶
An example of functional group removal (FGR) is shown below. The two phenyl groups are temporarily removed from the target structure, and are introduced in due time with a Grignard reaction. The synthesis is based on simple starting material such as ethyl acrylate, piperidine and bromobenzene (for the Grignard).

The Retrosynthetic Process¶
The retrosynthetic process has often been compared to a game of chess. Organic synthesis is a science and a fine art in which masters such as Corey, introduced logical rules that are now the canon of every undergraduate synthetic chemistry course. The process starts by recognizing the key functional groups of the target compound and their possible transformations (e.g. FGA, FGI and FGR). Strategic bonds are then disconnected and assessed to see whether they lead to simple starting material.

From TC to SM¶
Disconnecting in the middle of the molecule or at branch points from chains may yield useful simplifications. After locating the functional groups and their relationships all C-X and C-C bonds are then systematically disconnected, until available starting material is found. Retrosynthetic schemes provide useful material for thinking about ways precursors can be prepared and whether they correspond to reliable reactions. The chemistry associated to a disconnection is usually obvious; however in the intermediate steps the transformations of precursors may require more creativity.

articles
Development of a Scalable Process for an Endothelin Antagonist, CI-1034 Thomas E. Jacks 25th Symposium on Synthetic Organic Chemistry, Holland, Michigan
May 31, 2002
The Synthetic Program¶
As long as a retrosynthetic pathway does not end up with accessible starting material, the analysis is worth nothing. The most promising scheme finally adopted represents "the project": it serves to define phases, milestones and work plans for the group of chemists involved in the synthetic program.

The Tetrodotoxin Example¶
The tetrodotoxin example is illustrated here; it shows the retrosynthetic scheme adopted by the Kishi group, and how the synthesis was developed.


articles
Synthetic studies on tetrodotoxin and related compounds. III. Stereospecific synthesis of an equivalent of acetylated tetrodamine Y. Kishi, M. Aratani, T. Fukuyama, F. Nakatsubo, T. Goto, S. Inoue, H. Tanino, S. Sugiura, and H. Kakoi J. Am. Chem. Soc. 94 1972
Synthetic studies on tetrodotoxin and related compounds. IV. Stereospecific total syntheses of DL-tetrodotoxin Y. Kishi, T. Fukuyama, M. Aratani, F. Nakatsubo, T. Goto, S. Inoue, H. Tanino, S. Sugiura, and H. Kakoi J. Am. Chem. Soc. 94 1972
Simple Exercise in Retrosynthesis¶
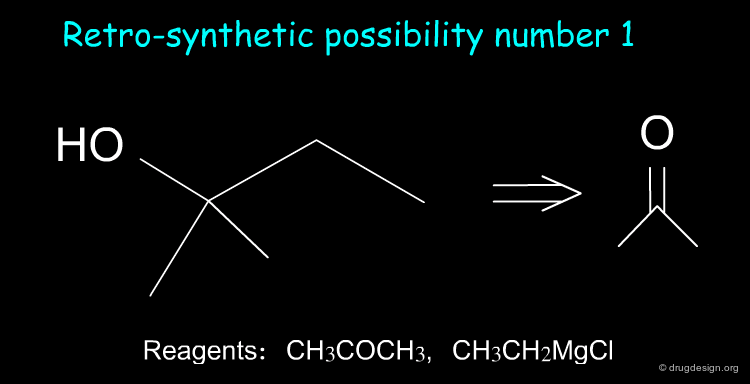
Suppose we want to synthesize the compound shown below, and in our laboratory we only have ethanol, acetone and all the inorganic reagents available. How do we proceed?

Retrosynthesis¶
Two retrosynthetic routes are feasible. We can use the reagents CH3COCH3 and CH3CH2MgCl, or, alternatively, CH3COCH2CH3 and CH3MgCl. We choose the first option because butanone is not available in the laboratory.

Synthesis¶
The synthesis based on the chosen retrosynthetic route adopted can then be implemented. Starting with ethanol the desired compound is obtained as follows.

Disconnection Methods for Retrosynthesis¶
The disconnection method plays a central role in retrosynthetic analyses. For a drug to be prepared, there are three ways to break or make a bond which can be used in retrosynthetic analyses. These are known as the homolytic, the heterolytic and the pericyclic disconnections.

Homolytic Disconnection¶
Homolytic fission is a free radical disconnection. Free radical disconnections are generally not very useful in the synthesis of novel drugs because the reconnection reaction proceeds through free radical mechanisms, which usually produce mixtures and are not desirable in the synthesis of a drug.

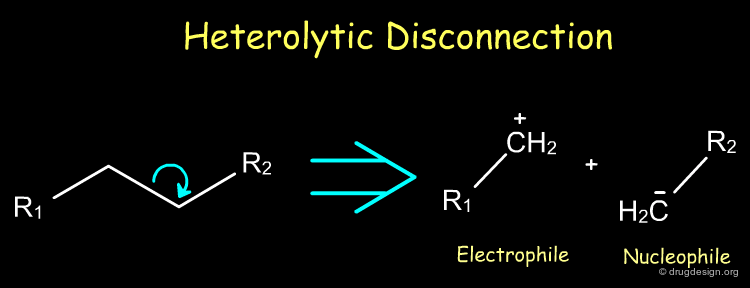
Heterolytic Disconnection¶
Heterolytic disconnections are very useful because they result in the formation of electrophile and nucleophile species. Such species are synthons that can be converted into a reagent for the initiation or the propagation of a synthetic scheme.
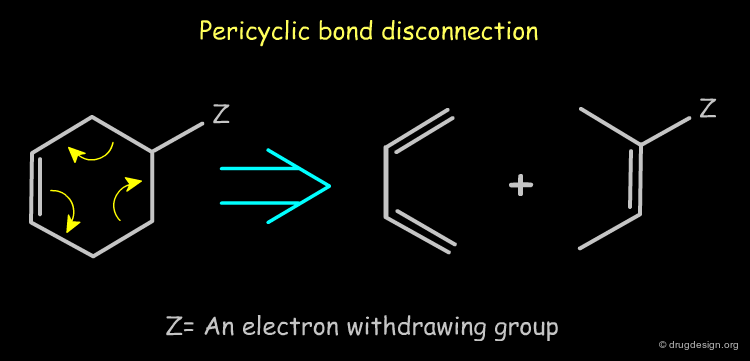
Pericyclic Bond Disconnection¶
Rings that carry an electron-withdrawing group can be disconnected by a reverse pericyclic mechanism (i.e. Diels-Alder). Such disconnections are useful in synthetic medicinal chemistry because they lead to synthons that can be converted to reagents.
Combining the Three Synthetic Methods¶
A synthesis can be based on existing knowledge, on existing material, or on creative design. A good synthetic route is generally an intelligent combination of all three, incorporating known and reliable chemical reactions, readily available compounds, and a good dose of ingenuity.
Synthetic Tactic¶
Principles of Synthetic Tactics¶
Once the global strategy has been defined, a plan of attack is formulated, which defines specific actions to achieve the goal. These actions consist of a set of maneuvers, each of which requires adequate available resources to achieve the ultimate goal.

Reaction Classifications¶
One of the best ways to organize a plan of attack is to use operational classifications of chemical reactions. There are different ways of characterizing organic reactions, which are useful tools for recognizing patterns of chemical reactivity. These are crucial to the design of a reaction. Some of these classifications are presented in the following pages.

By Changes Occurring in the Reactant Molecules¶
When considering the structural changes occurring in the reactant molecules, it is useful to distinguish broad classes of reactions such as addition, elimination, substitution and rearrangements.

By Reaction Types¶
It is sometimes useful to describe reactions in terms of changes of properties (e.g. oxidation state, acidity etc...) or in terms of species that seek out charged centers (e.g. nucleophiles, electrophiles). In the example below, we look at the oxidation changes of the carbon atoms of the double bond: one has been oxidized (C2) and the other (C3) reduced. Note that this type of description is independent of the mechanism.
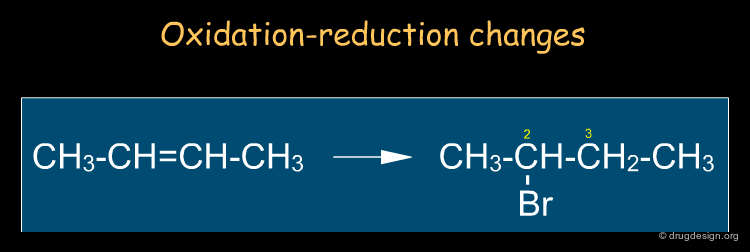
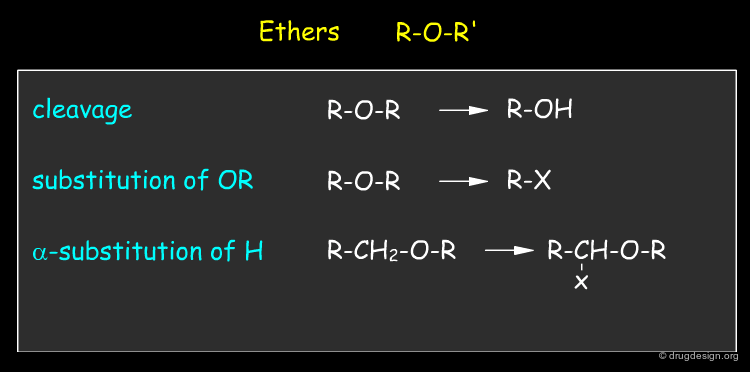
By Functional Groups¶
Functional groups characterize the reactivity of molecules. Since most reactions occur at, or adjacent to functional groups, it is useful to identify them in terms of chemical functionality. Below are given some examples of functional groups with their most characteristic reactions.





Protecting Groups¶
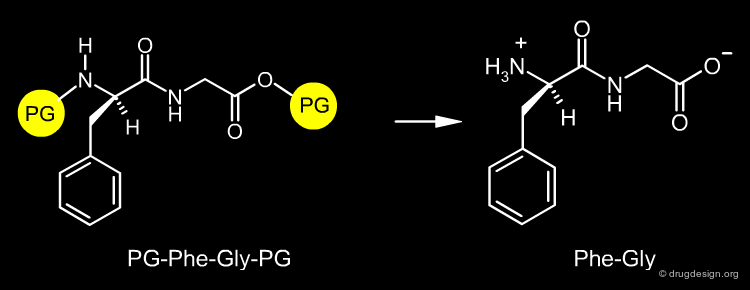
The design of a synthetic pathway often requires a reaction to be carried out at a certain center of the molecule. However, another center may interfere and undergo a similar undesired reaction. The synthetic medicinal chemist must find a way to protect the one center and allow the reaction to occur at the other one. For example if the dipeptide Phe-Gly is needed, it can be prepared by the condensation of the two amino-acids Phe and Gly; however if these amino-acids are not properly protected, the reaction can produce a mixture of the Phe-Gly, Phe-Phe, Gly-Phe and Gly-Gly dipeptides.

Protecting Reactive Centers¶
Protecting groups are necessary to protect a reactive center that at a certain step should not be modified. Their role is to mask the particular center in order to allow a transformation to be carried out elsewhere in a molecule. In our example, the Phe-Gly dipeptide can be obtained quantitatively, by protecting the amino of the phenylalanine (N-protection) and the carboxyl of the glycine (O-protection).

Requirements for Protecting Groups¶
The protecting groups must have the following requirements: (a) easy to attach to the appropriate functional group; (b) form a stable structure that is not affected by the reaction conditions and reagents; (c) easy to remove once it is not required; (d) can be converted into another structure as part of the synthesis.
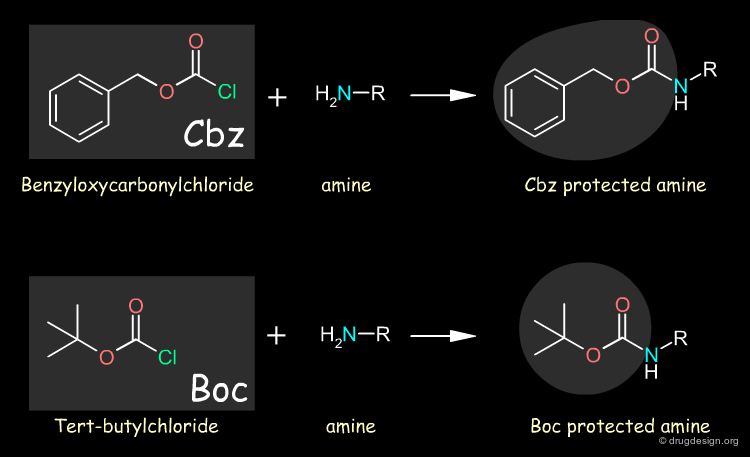
Protection and Deprotection of Amino Acids¶
Amino groups are normally protected by converting them to amides whereas carboxyl groups are converted to esters. Examples of amino protecting groups are benzyloxycarbonyl (abbreviated as Z or Cbz) and tert-butyloxycarbonyl (Boc) that are introduced using the chloride. When needed, the protecting group can be easily removed: Cbz can be removed with catalytic hydrogenolysis under very mild conditions or HBr in acetic acid; and Boc can be removed with trifluoroacetic acid in methylene chloride or HBr in acetic acid.


Examples of Protecting Groups¶
A variety of protecting groups for a certain functional group can be used and then removed at the intermediate or final steps of the synthesis. Some examples are presented in the following pages.

Alcohol Protection¶
Several groups can be used for alcohol protection such as triphenylmethyl (trityl) acetate or trifluoroacetate. These groups may need different conditions for their removal: for example, trityl needs acid conditions while acetate or trifluoroacetate need base conditions.

Carboxylic Acid Protection¶
The carboxylic group is one of the most common groups that must be protected during synthesis. Two examples of protecting groups and corresponding removal reagents are given below.

Amine Protection¶
Amine groups are often present and need to be protected properly. In particular peptide synthesis requires their selective protection and removal.

Phenol Protection¶
The hydroxyl group of alcohols and phenols frequently requires protection. The benzyl group is a suitable protecting group which can be removed by catalytic hydrogenation.

How to Assess the Quality of a Synthesis?¶
There is no single correct pathway to the synthesis of a molecule. Some routes, however, are better than others. Measurable means for assessing the quality of a synthesis exist and include: the number of steps, good selectivity, good yields, economy of raw material and mild reaction conditions. Other factors are difficult to quantify but are also important, such as the creativity and elegance, practicality and convenience of the steps, flexibility with respect to problems, lack of involvement of toxic compounds or laborious purification procedures.

Syntheses of Swainsonine¶
Swainsonine illustrates how a molecule can be synthesized by many different routes. Look at the corresponding articles and rank them in terms of your own preference.





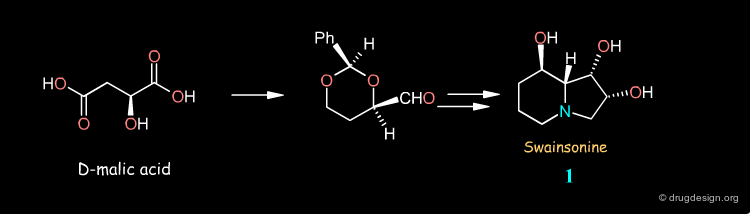

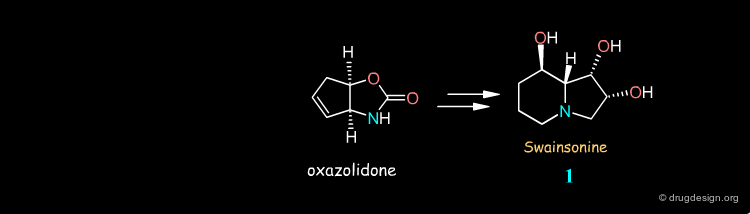
articles
Asymmetric synthesis of (-)-swainsonine, (+)-1,2-di-epi-swainsonine, and (+)-1,2,8 -tri-epi-swainsonine. Lindsay KB, Pyne SG. J Org Chem. 67(22) 2002
Synthesis of the new mannosidase inhibitors, diversity-oriented 5-substituted swainsonine analogues, via stereoselective Mannich reaction. Fujita T, Nagasawa H, Uto Y, Hashimoto T, Asakawa Y, Hori H. Org Lett. 6(5) 2004
Stereoselective synthesis of swainsonines from pyridines Gerres Heimgaertner, Dirk Raatz and Oliver Reiser Tetrahedron 61 2005
Synthesis of the Novel Mannosidase Inhibitors (3R)- and (3S)-3-(Hydroxymethyl) swainsonine Erik J. Hembre and Willimn H. Pearson Tetrahedron 53 1997
A New Approach to Indolizidine Alkaloids: Asymmetric Formal Total Synthesis of (-) -Swainsonine Wei-Shan Zhou, Wen-Ge Xie, Zhi-Hui Lu, Xin-Fu Pan Tetrahedron Letters 36, No. 8 1995
Alpha-Acylamino Radical Cyclizations: Application to the Synthesis of (-) - Swainsonine Jeffrey M. Dener, David J. Hart, and Subban Ramesh J. Org. Chem. 53, No. 26 1988
New Chiral Route to (-)-Swainsonine via an Aqueous Acylnitroso Cycloaddition Approach Masaichi Naruse, Sakae Aoyagi, and Chihiro Kibayashi J. Org. Chem. 59 1994
Diastereoselective synthesis of new polyhydroxylated indolizidines from (L)- glutamic acid Katarina Kadlecikova, Vincent Dalla, Stefan Marchalin, Bernard Decroix and Peter Baranc Tetrahedron 61 2005
Synthesis of oxygen- and nitrogen-containing heterocycles by ring-closing metathesis. Alexander Deiters and Stephen F. Martin Chem Rev. 104(5) 2004
An Enantiocontrolled Synthesis Of (-)-Swainsonine Sung Ho Kang and Geun Tae Kim Tetrahedron Letters 36, No. 28 1995
Synthesis and mannosidase inhibitory activity of 3-benzyloxymethyl analogs of swainsonine William H. Pearson and Luyi Guob Tetrahedron Letters 42 2001
Stereochemical Issues¶
Why Chiral Drugs?¶
Biological receptors are chiral and involve a high degree of stereoselectivity. Chiral molecules are often used in drug research because of their 3D molecular arrangement, which is particularly adapted for meeting the stereochemical requirements of the receptor.

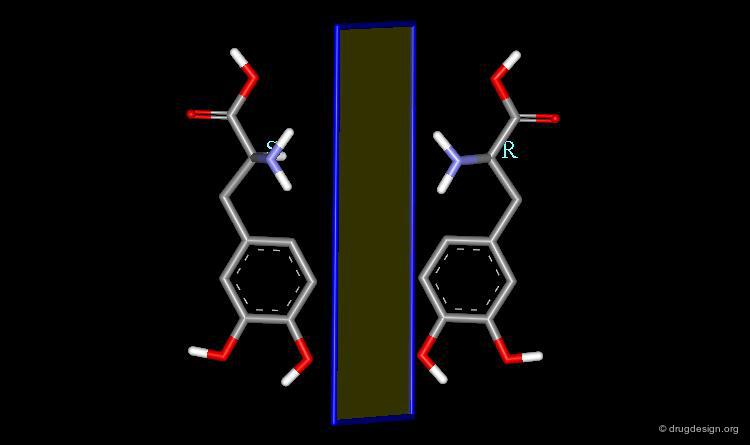
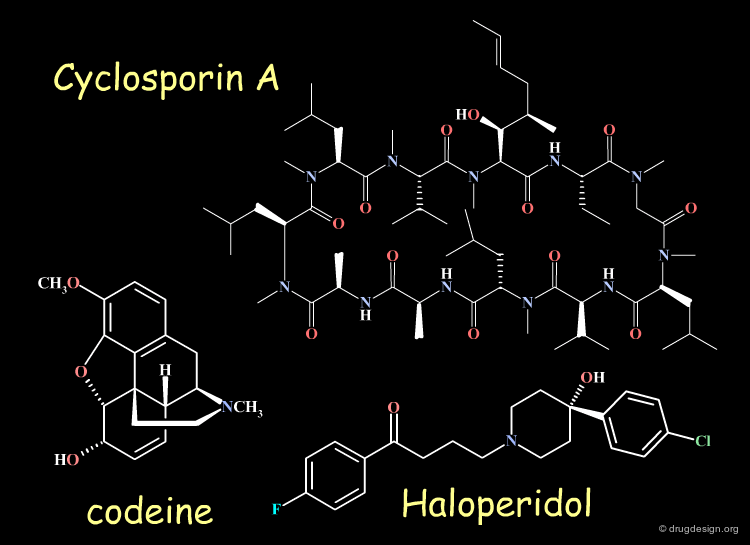
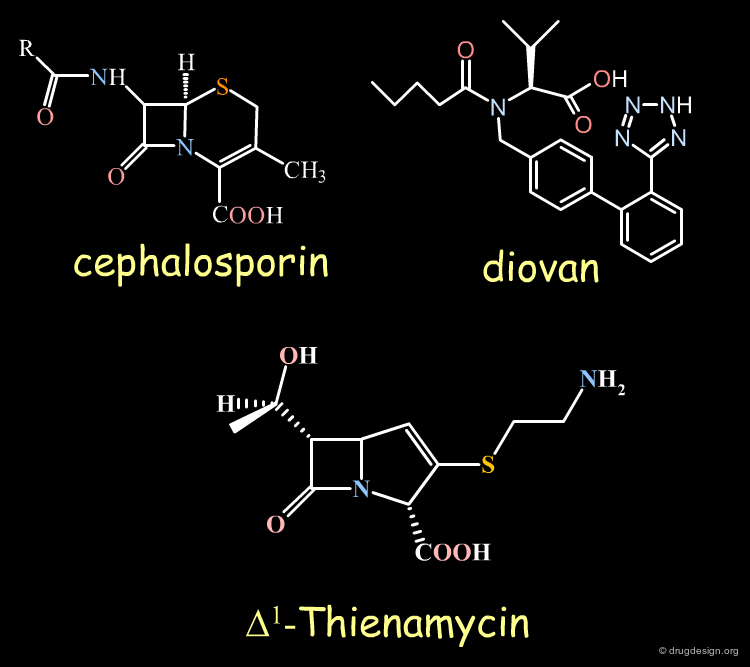
Examples of Chiral Drugs¶
Many drugs are chiral. In fact, over 50% of the top-selling 500 pharmaceutical drugs on the market in 2004 were chiral molecules. Examples of some well known chiral drugs are shown below.



Multiple Aspects of Molecular Chirality¶
The stereochemical course of a reaction is influenced by factors such as reactant and transition state energies, as well as the conformational and electronic properties. These properties are extensively presented in other chapters of this course, and the reader is invited to review them for more detail. In the following pages we focus on synthetic methods for preparing chiral molecules.

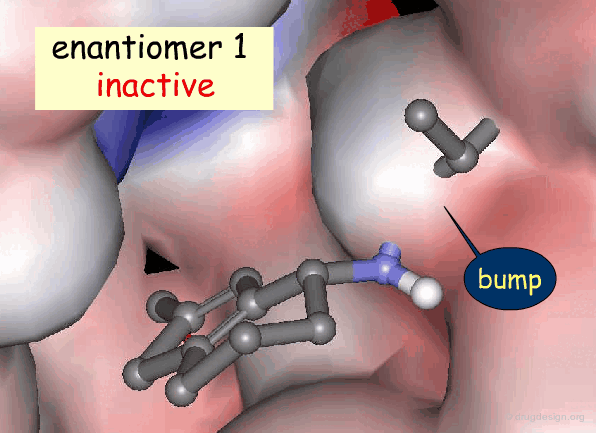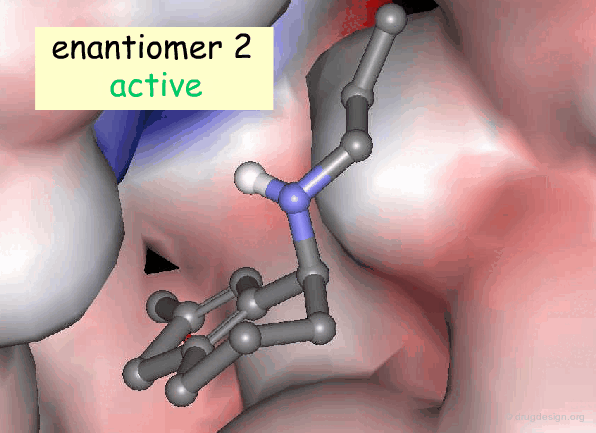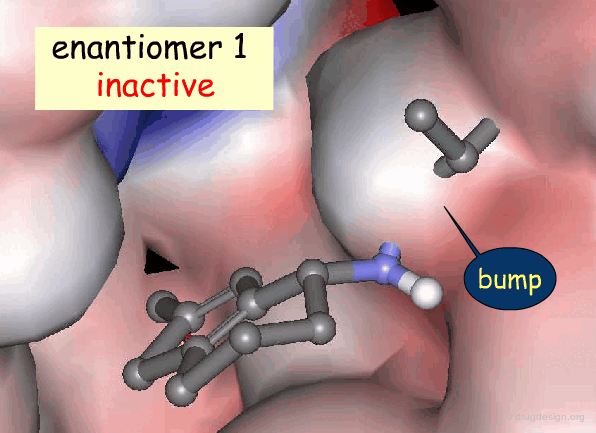
Active and Inactive Enantiomers¶
As a consequence of the difference in molecular shape of enantiomers and the high degree of chirality of biological receptors, only one enantiomer is generally the active principle.
'Chiral Switch' : From a Racemic to a Chiral Drug¶
In the late 1980s, most chiral drugs were commercialized as racemic mixtures. In many cases, one of the enantiomers was found to have more favorable therapeutic and pharmacokinetic characteristics only after commercialization of the racemate. A new trend known as 'chiral switching' has recently emerged, which aims at the development of single enantiomers of commercial racemate drugs. Since 1990, the proportion of single-enantiomer drugs approved as new chemical entities has been consistently greater than that of racemates. Today, market approval of a racemate has become much more stringent.

articles
The Chiral Switch: The Development Of Single Enantiomer Drugs From Racemates Hutt, A.J. and Valentova, J. Acta Facult. Pharm. Univ. Comenianae 50 2003
Advantages of Single Enantiomer Drugs¶
The advantages of single enantiomer drugs are multiple. These involve its pharmacokinetic properties, selectivity and patent related aspects, all of which are detailed in the following pages.

Pharmacokinetic Properties¶
Because of their different 3D configurations, the pharmacodynamic and pharmacokinetic properties of enantiomers are likely to be different. When this is found to be true and one of the enantiomers has greater therapeutic and pharmacokinetic characteristics, a single enantiomer drug presents health benefits including an improved safety margin and other advantages such as those indicated below.

Perhexiline¶
Perhexiline (an anti-angina drug), is an example of the different pharmacokinetic properties of enantiomers. The racemic of perhexiline was withdrawn from the market because it killed individuals in whom the wrong enantiomer was more slowly metabolized and who accumulated gram quantities of it. If the developers of the racemate had been aware of this noxious effect, they would have marketed a drug with only the fast-clearing enantiomer.

Selectivity¶
When one enantiomer is responsible for the activity of interest, its paired 'inactive' enantiomer also has properties and effects that may not be negligible. It can interact with other receptors, cause side effects and be toxic. Some examples to illustrate this are given in the following pages.

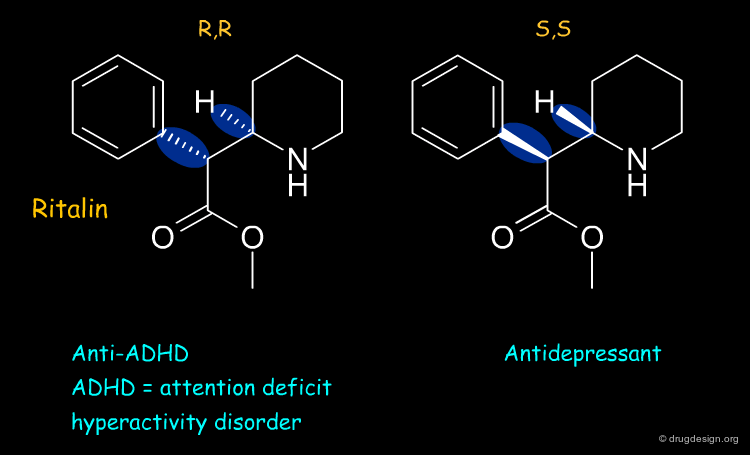
Ritalin¶
Ritalin is used to treat attention deficit hyperactivity disorder (ADHD). The (R,R) enantiomer of ritalin has the anti-ADHD effect whereas the (S,S) form acts as an antidepressant.
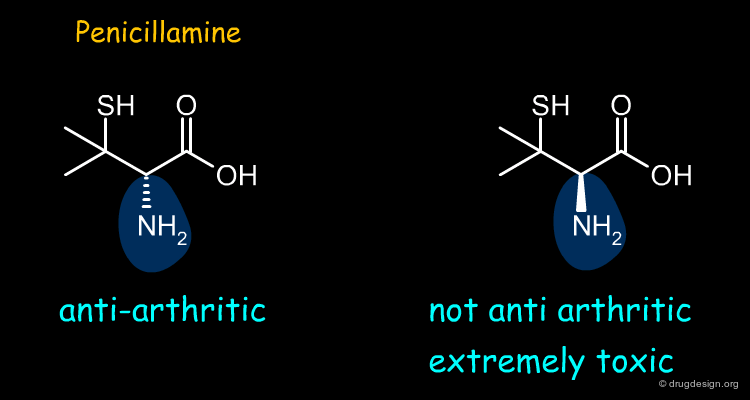
Penicillamine¶
Penicillamine is an anti-arthritic drug. Most of the anti-arthritic activity of penicillamine resides in the (S)-enantiomer whereas the (R) form is extremely toxic.
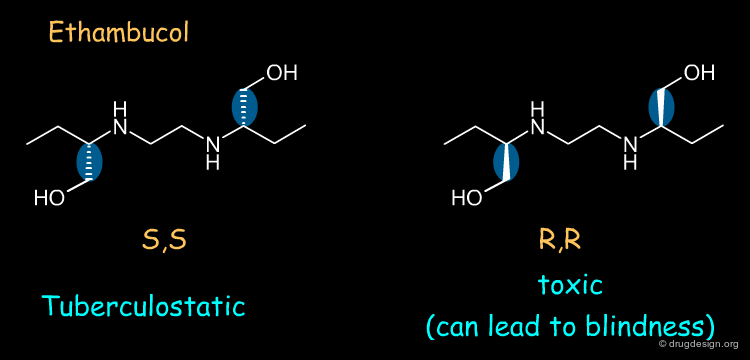
Ethambutol¶
Ethambutol is a tuberculostatic drug. The (S,S) enantiomer of ethambutol is a tuberculostatic agent whereas the (R,R) enantiomer is toxic (causes optical neuritis that can lead to blindness).
Ketamine¶
Ketamine is an anesthetic drug. The S-enantiomer of ketamine possesses the anesthetic activity; the R-enantiomer can cause hallucinations.

L-Dopa¶
Levodopa (L-dopa) is an anti-Parkinsonian drug that is commercialized in an enantiomerically pure form because the D-form is toxic and causes serious side effects (such as granulocytopenia).

Patent Position¶
Chiral switching can be a useful marketing strategy for a company aiming at extending its patent franchise and interested in protecting itself from generic companies. For example the AstraZeneca patent for the ulcer drug Prisolec expired in 2002; the company successfully launched Nexium in 2001, the S-enantiomer of Prisolec. This enabled the company to keep its share of the market intact.

Three Methods to Obtain Chiral Molecules¶
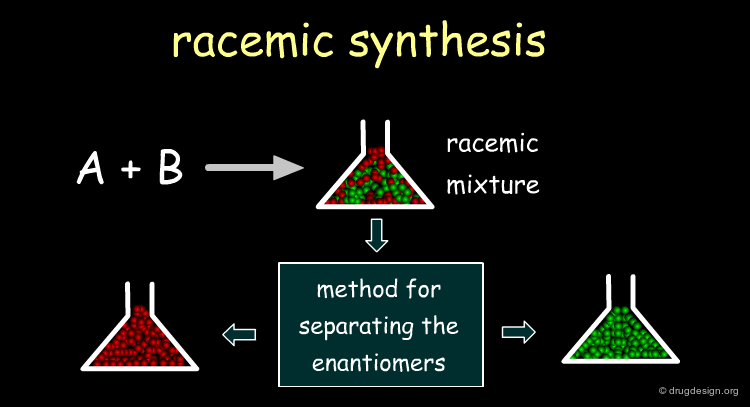
There are three approaches of obtaining a chiral molecule. The first consists of the synthesis of the racemate, followed by the separation of the enantiomers. The second is an asymmetric route for preparing the molecule with the proper stereochemistry. The third approach consists of extracting the chiral principle from natural sources (plants, animals).

Racemic Route¶
It is usually simpler to develop a racemic synthesis than an asymmetrically pure one, and afterwards find a method for separating the enantiomers. In drug discovery it is crucial to rapidly obtain pure enantiomers for pharmacological tests, and the racemic route is preferred. Moreover, resolution methods enabling access to both enantiomers are also useful when preclinical studies are required for each enantiomer.
articles
Asymmetric Catalysis in the Pharmaceutical Industry Joel M. Hawkins, Timothy J. N. Watson Angew. Chem. Int. Ed. Engl. 43, Issue 25 2004
Development of the Commercial Process for Zoloft/Sertaline George J. Quallich Chirality 17 2005
Resolution of Racemates¶
In the mid 1980s enantiomers were separated by first forming salts or derivatives with chiral reagents, and then resolving them by conventional methods such as crystallization or chromatography on silica. Chromatography methodologies have now become the most widely utilized approach for the resolution of racemates. New developments such as countercurrent, supercritical fluid and simulated moving bed continuous chromatography have begun to revolutionize the drug development process in the industry.

Example of Industrial Racemate Separation¶
Zoloft (compound 4), also known as sertraline is a chiral antidepressant drug marketed as a 1S(+),4S(+) enantiomer. Its synthesis is based on the key chiral tetralone 2. This building block is prepared industrially with high purity and yield from racemate 1, using simulated moving bed (SMD) continuous chromatographic technologies and racemization of the undesired enantiomer.
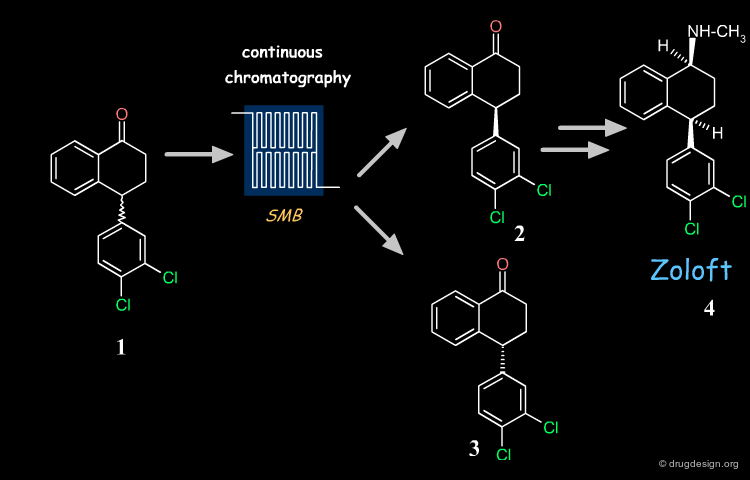
Advantages of the Process Chosen¶
The industrial synthesis currently used for Zoloft eliminated the need for reagents such as titanium chloride and D-mandelic acid used in the initial inefficient route, and also removed the uncertainties surrounding royalties for particular reagents introduced by the medicinal chemists. It saved solvents, reagents and plant capacity previously harnessed for processing the undesired enantiomer, and resulted in immediate cash savings for Pfizer.

Recycling Undesired Enantiomer¶
If the undesired enantiomer can be re-racemized, this enables all the racemate to be progressively converted into the desired isomer. There are many industrial examples of such recycling. One example is the tetralone intermediate involved in the synthesis of Zoloft whose unwanted enantiomer is repeatedly recycled, as visualized below. With this procedure the desired enantiomer is prepared industrially with high purity and yield (ee = 99%; recovery yield 98%).

articles
Development of the Commercial Process for Zoloft/Sertaline George J. Quallich Chirality 17 2005
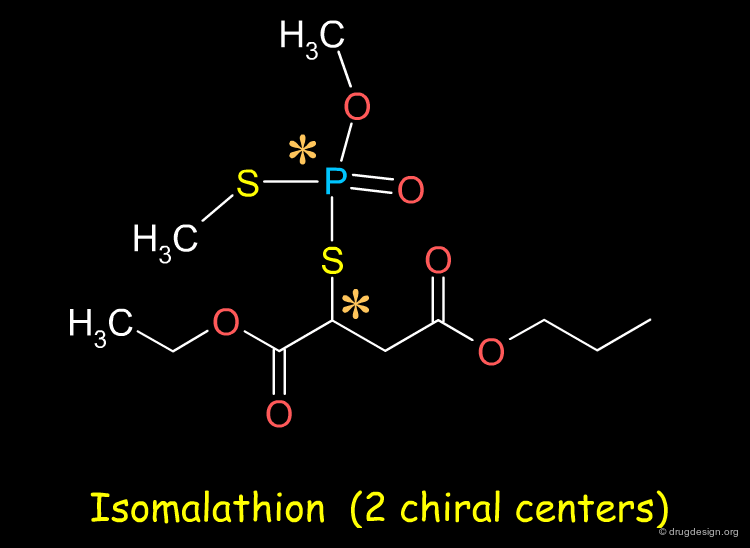
Sometimes Separation very Difficult¶
The separation of enantiomers can be very challenging. For example the resolution of the four stereoisomers of the organophosphoramidate compound isomalathion is a difficult problem: for some compounds it may not be straightforward to separate the enantiomers.
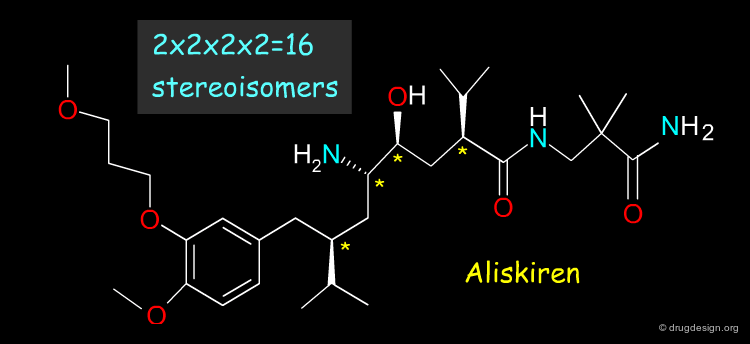
When Too Many Chiral Centers¶
The racemic route is not realistic when the molecule contains too many chiral centers; for example Aliskiren (a renin inhibitor now in late stages of development) has 4 asymmetric centers. For molecules with many chiral centers a stereoselective route is necessary.
Asymmetric Route¶
Asymmetric synthesis aims at the direct preparation of the target molecule with the proper chirality. Brilliant molecule builders such as Woodward, Corey, Kishi, Nicolaou and others succeeded in synthesizing complicated chiral molecules with intelligent science and fine art. Thanks to their heroic efforts, an expanding arsenal of tools is now available in synthetic chemistry for the preparation of chiral molecules. Chirality can be introduced either through chiral reagents or chiral catalysts.

Chiral Reagents and Reactants¶
Aliskiren is shown here to illustrate the asymmetric synthesis of a drug. Lactone 2 is a key building block; its opening gives the proper stereochemistry for both the hydroxyl and the isopropyl groups located on the right side of the structure. Likewise the chirality of the second isopropyl and that of the amino are also introduced stereospecifically (not shown here). All four chiral centers of Aliskiren are created by asymmetric syntheses.

articles
The Power of Visual Imagery in Synthesis Planning Stereocontrolled Approaches to CGP-60536B, a Potent Renin Inhibitor S. Hanessian, S. Claridge, and S. Johnstone J. Org. Chem. 67 2002
A Convergent Synthesis of the Renin Inhibitor SPP-100 Using a Nitrone Intermediate Alessandro Dondoni, Geert De Lathauwer and Daniela Perrone Tetrahedron Letters 42 2001
A Convergent Synthesis of the Renin Inhibitor CGP60536B David A. Sandham, Roger J. Taylor, John S. Carey and Alexander Faessler Tetrahedron Letters 41 2000
A Convergent Synthesis Approach Towards CGP60536B, a Non-Peptide Orally Potent Renin Inhibitor, Via An Enantiomerically Pure Ketolactone Intermediate Heinrich Rueeger, Stefan Stutz, Richard Goeschke, Felix Spindler and Juergen Maibaum Tetrahedron Letters 41 2000
Novel 2,7 Dialkyl Substituted 5(S)-Amino-4(S)-Hydroxy-8-Phenyl-Octanecarboxamide Transition State Peptidomimetics are Potent and Orally Active Inhibitors of Human Renin R. Goeschke, V. Rasetti, N.C. Cohen, J. Rahuel, M. Gruetter, S. Stutz, H. Rueger, J.M. Wood, J. Maibaum XVth Meeting of the European Federation of Medicinal Chemistry, Edinburg
Sept. 6- 10, 1998
Design and Syntheses of Novel 2,7Dialkyl substituted 5(S)Amino 4(S)Hydroxy-8Phenyl- Octanecarboxamides-as in vitro Potent Peptidomimetic Inhibitors of Humain Renin R. Goeschke, N.C. Cohen, J.M. Wood and J. Maibaum Bioorganic and Med. Chem. Letters 7 1997
Chiral Catalysts¶
Chiral catalysts such as metal complexes or enzymes can be used to create new asymmetric centers starting from prochiral molecules. In the example below the painkiller S-Naproxen is obtained with good yield and purity using Ru-S-BINAP as a catalyst for the hydrogenation.

Asymmetric Synthesis in R&D.¶
The development of an asymmetric synthesis is generally not considered in the early drug discovery stage, since it is both time-consuming and expensive: whenever possible the racemic route is preferred. For large scale production of pure stereo-isomers, the set up and optimization of an asymmetric process still requires substantial experimentation and expensive catalysts, with the important constraint of being economically acceptable.

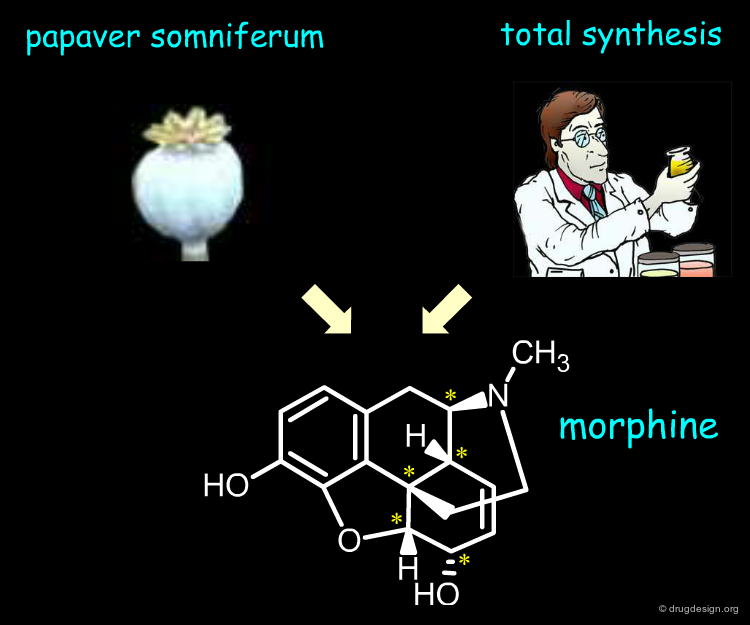
Extraction From Natural Sources¶
Besides the therapeutic advantages of single enantiomers, their industrial manufacturing is an important issue. For example pain relieving morphine has five asymmetric centers. The molecule is synthesized by the poppy plant Papaver somniferum. On the other hand Woodward and other groups successfully achieved the total synthesis of this alkaloid. However, the costs associated with the preparation of large amounts are such that the extraction approach is by far the more economical.
articles
Synthesis of (-)-Morphine Douglass F. Taber, Timothy D. Neubert, and Arnold L. Rheingold J. Am. Chem. Soc. 124 2002
The Synthesis of Morphine M. Gates, G. Tschudi J. Am. Chem. Soc. 74 1952
The Synthesis of Morphine M. Gates, G. Tschudi J. Am. Chem. Soc. 78 1956
The Synthesis of Morphine D. Elad, D. Ginsburg J. Am. Chem. Soc. 76 1954
Biogenetically patterned synthesis of the morphine alkaloids M. A. Schwartz, I. S. Mami J. Am.Chem. Soc. 97 1975
Synthetic opium alkaloids and derivatives. A short total synthesis of (+-)- dihydrothebainone, (+-)-dihydrocodeinone, and (+-)-nordihydrocodeinone as an approach to a practical synthesis of morphine, codeine, and congeners K. C. Rice J. Org. Chem. 45 1980
Studies Directed Towards The Total Synthesis Of Morphine Alkaloids D. A. Evans, C. H. Mitch Tetrahedron Lett. 23 1982
Synthesis via vinyl sulfones. 21. Total synthesis of dl-morphine J. E. Toth, P. L. Fuchs J. Org. Chem. 52 1987
Convergent synthesis of (+-)-dihydroisocodeine in 11 steps by the tandem radical cyclization strategy. A formal total synthesis of (+-)-morphine K. A. Parker, D. Fokas J. Am. Chem. Soc. 114 1992
Asymmetric synthesis of either enantiomer of opium alkaloids and morphinans. Total synthesis of (-)- and (+)-dihydrocodeinone and (-)- and (+)-morphine C. Y. Hong, N. Kado, L. E. Overman J. Am. Chem. Soc. 115 1993
Formal Total Synthesis of ( - )-Morphine by Cuprate Conjugate Addition J. Mulzer, G. Duerner, D. Trauner Angew. Chem. Int. Ed. Engl. 35 1996
Stereoselectivity of Action not Always Predictable¶
Although there are theoretical advantages in developing single enantiomer drugs, there are cases where the approach has failed. For example Dilevalol was thought to have advantages over the corresponding racemic drug Labetalol (a beta-blocker launched in 1977), but it was taken off the market because of toxicity. ADME/Tox is a science that is far from being fully understood; the pharmacokinetic and pharmacodynamic data of racemic mixtures and enantiomers can be ambiguous and mask unpredictable effects.

Spontaneous Enantiomer Interconversion¶
Normally a stereospecific synthesis aims at obtaining the desired active enantiomer. Thalidomide is a special case where such effort was worthless. The drug was introduced in the 1950s as a sedative and was used by pregnant women to treat morning sickness. Birth defects were observed and Thalidomide was banned worldwide. Only the R enantiomer has the sedative action, whereas the S enantiomer was found to be teratogenic. However the active enantiomer racemizes in less than 10 minutes in the body and the tragedies could not have been avoided by use of the active R-form.

Structural Diversity and Higher Level Synthetic Strategies¶
Diverse Strategy in Drug Discovery¶
The space of synthesizable molecules is huge. Structural diversity is essential for lead discovery, and the generation of a great number of diverse molecules increases the chances of finding novel compounds with significant therapeutic value.

Introducing Diversity in the Reagents¶
Diversity is always introduced by the reagents. In the example below, diverse compounds such as 1 need to be prepared. They are synthesized by the reaction of building block 2 with a number of diverse reagents that can be either purchased or simply prepared.

Introducing Diversity at the Proper Time¶
The introduction of diversity through reagents should be made at the right time. Important considerations include when protection/deprotection of functional groups should take place and fully exploit the strength of the synthetic route, whether it is linear or convergent.
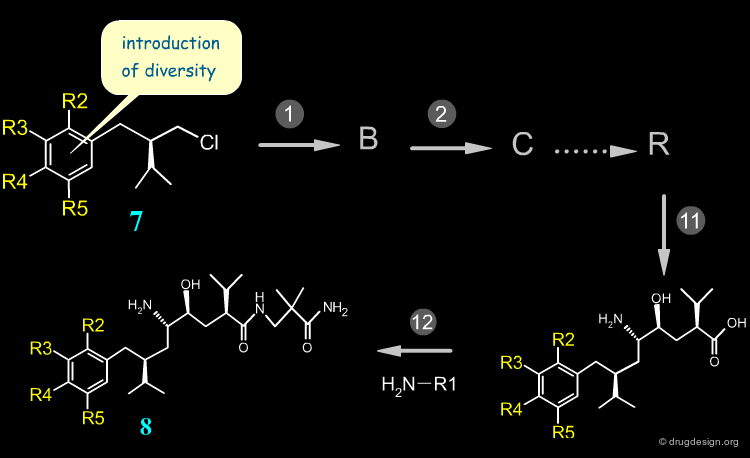
Late Stage Introduction of Diversity¶
When variations are introduced at the end of the synthesis, many diverse analogs can be prepared in a minimum number of steps. This is the case of Aliskiren that was synthesized using the retrosynthetic scheme shown here (button "Retrosynthesis"). Hundreds of amide analogs such as 6 were prepared from acid 2 in order to optimize the series.


articles
The Power of Visual Imagery in Synthesis Planning Stereocontrolled Approaches to CGP-60536B, a Potent Renin Inhibitor S. Hanessian, S. Claridge, and S. Johnstone J. Org. Chem. 67 2002
A Convergent Synthesis of the Renin Inhibitor SPP-100 Using a Nitrone Intermediate Alessandro Dondoni, Geert De Lathauwer and Daniela Perrone Tetrahedron Letters 42 2001
A Convergent Synthesis of the Renin Inhibitor CGP60536B David A. Sandham, Roger J. Taylor, John S. Carey and Alexander Faessler Tetrahedron Letters 41 2000
A Convergent Synthesis Approach Towards CGP60536B, a Non-Peptide Orally Potent Renin Inhibitor, Via An Enantiomerically Pure Ketolactone Intermediate Heinrich Rueeger, Stefan Stutz, Richard Goeschke, Felix Spindler and Juergen Maibaum Tetrahedron Letters 41 2000
Novel 2,7 Dialkyl Substituted 5(S)-Amino-4(S)-Hydroxy-8-Phenyl-Octanecarboxamide Transition State Peptidomimetics are Potent and Orally Active Inhibitors of Human Renin R. Goeschke, V. Rasetti, N.C. Cohen, J. Rahuel, M. Gruetter, S. Stutz, H. Rueger, J.M. Wood, J. Maibaum XVth Meeting of the European Federation of Medicinal Chemistry, Edinburg
Sept. 6- 10, 1998
Design and Syntheses of Novel 2,7Dialkyl substituted 5(S)Amino 4(S)Hydroxy-8Phenyl- Octanecarboxamides-as in vitro Potent Peptidomimetic Inhibitors of Humain Renin R. Goeschke, N.C. Cohen, J.M. Wood and J. Maibaum Bioorganic and Med. Chem. Letters 7 1997
Early Stage Introduction of Diversity¶
When variations are needed far from the end of the synthesis, diverse analogs are very difficult to prepare. This is the case of the aryl moiety of Aliskiren. Many reagents of structure 7 must be prepared and moreover, additional reactions are necessary before arriving at the target compounds 8. With this synthesis the preparation of many diverse aryl derivatives is extremely difficult.
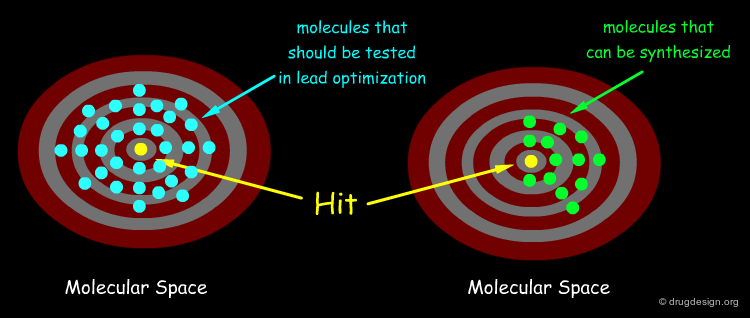
Diversity Space Restricted by Synthetic Route¶
As chemistry is based on the transformation of functional groups, inevitably a given synthetic scheme restricts the field of the possible diverse analogs that can be synthesized. In theory, several reaction routes are needed to better cover the entire diversity space, but in practice this is not always feasible. This is discussed in the following pages.
Consider Alternative Syntheses¶
One way of escaping the bias of a single synthetic scheme is to consider multiple routes. This may be important because a single change in a fragment may transform a micromolar hit into a nanomolar one. Very often the optimization of a lead is greatly restricted, the molecules prepared are not those that should be made and the optimization process is trapped with no substantial change in biological activities. Another synthesis is likely to open up new possibilities and reveal an entirely different moiety that can lead to potency and selectivity.

Example of Introduction of Diversity¶
With the aim of incorporating diversity in a structure, it may be necessary to modify the initial lead. In the example below the Abbott group replaced the indane moiety of a SmithKline lead by a pyrrolidine. This novel structure permitted the synthesis of many analogs and the rapid generation of useful structure-activity relationships.

articles
2,4-Diarylpyrrolidine-3-carboxylic acids--potent ETA selective endothelin receptor antagonists. 1. Discovery of A-127722.
Winn M, von Geldern TW, Opgenorth TJ, Jae HS, Tasker AS, Boyd SA, Kester JA, Mantei RA, Bal R, Sorensen BK, Wu-Wong JR, Chiou WJ, Dixon DB, Novosad EI, Hernandez L, Marsh KC.
J Med Chem.
39(5) 1996
Combinatorial Chemistry¶
Combinatorial chemistry enables the creation of a large population of molecules (called libraries) that can be screened at one time. Typically, analogs of a common scaffold are synthesized in an automated manner (robotics) with a common synthetic route. Examples of libraries that can be created with these technologies are visualized below. By producing large and diverse compound libraries, the chemist increases the probability of finding novel compounds of interesting therapeutic value. The field of combinatorial chemistry is presented in detail in a special chapter in this course.

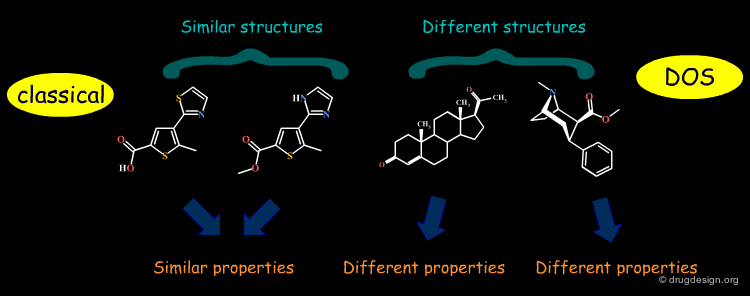
Diversity-Oriented Synthesis (DOS)¶
The synthetic strategy commonly used in combinatorial chemistry is to access diverse collections of small molecules by appending different sets of building blocks ("appendage decoration") to a common molecular skeleton. The impossibility of considering the scaffold as a "variable" has triggered the development of a new approach in synthetic chemistry known as "diversity-oriented synthesis" (DOS) which also aims at varying scaffolds.

Maximizing the Chance to Find a Hit¶
The quest for molecular diversity is based on the "similar property principle" which assumes that structurally similar molecules are likely to have similar properties and structurally dissimilar molecules are likely to have different properties. By enabling the variation of both scaffolds and substituents, DOS maximizes the chance of finding relevant hits.
Example of Diversity-Oriented Synthesis¶
An example of a DOS reaction is given below that shows how an immobilized dihydroisoquinoline reactive intermediate is used to obtain skeletally diverse compounds.

articles
Synthetic strategy toward skeletal diversity via solid-supported, otherwise unstable reactive intermediates. Taylor SJ, Taylor AM, Schreiber SL Angew Chem Int Ed Engl 43 2004
Higher Level Strategies in Organic Synthesis¶
The creation of more than one target molecule in a diversity-driven approach was introduced by Stuart Schreiber at Harvard University. Since the means available in synthetic chemistry are not suited to this approach, it has prompted many research groups to attempt to create new types of synthetic planning methods. The sketch shown below of the areas now covered by organic synthesis, suggests that the search for higher level strategies will be an important driving force for the next generation of methodology-oriented synthetic organic chemists.

articles
Target-Oriented and Diversity-Oriented Organic Synthesis in Drug Discovery Stuart L. Schreiber SCIENCE 287 17 March 2000
Generating Diverse Skeletons of Small Molecules Combinatorially Martin D. Burke, Eric M. Berger, Stuart L. Schreiber SCIENCE 302 24 October 2003
The small-molecule approach to biology Stuart L. Schreiber Chem Eng News 81 2003
A synthesis strategy yielding skeletally diverse small molecules combinatorially. Burke MD, Berger EM, Schreiber J Am Chem Soc 126 2004
A planning strategy for diversityoriented synthesis. Burke MD, Schreiber SL Angew Chem Int Ed Engl 43 2004
Diversity-Oriented synthesis of biaryl-containing medium rings using a one bead/one stock solution platform. Spring DR, Krishnan S, Blackwell HE, Schreiber SL J Am Chem Soc 124 2002
Synthesis of Some Common Drugs¶
Introduction to Synthetic Schemes¶
In the previous pages the main principles that govern the synthesis of novel drugs were discussed. In this section real examples are presented and include the following information: the chemical formula of the drug, a retrosynthetic analysis, the synthesis and some physico-chemical properties of the drug. We start with benzocaine.

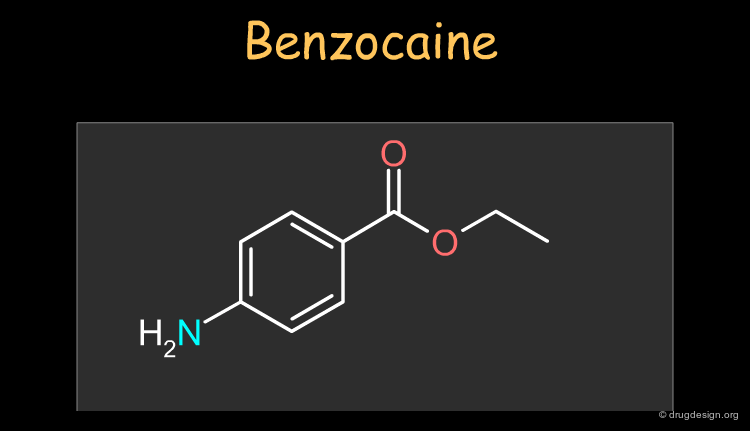
Benzocaine¶
Benzocaine is a widely used local and topical anesthetic. The drug can be synthesized as described in the following pages.
Retrosynthetic Scheme¶
Possible starting disconnections are the ester or the amine groups. The amine disconnection leads to toluene as starting material.
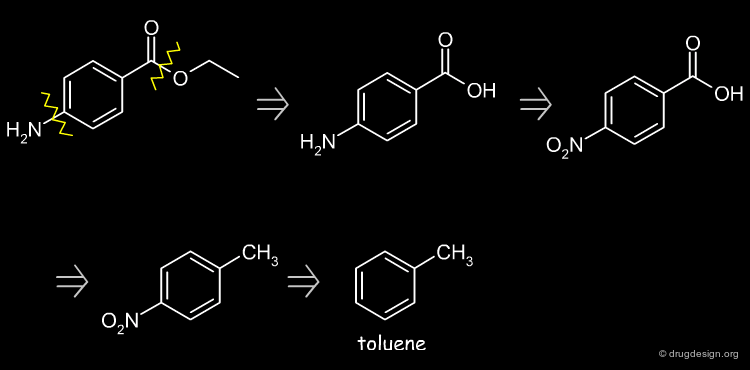
Benzocaine Synthesis¶
Nitration of toluene, oxidation of the methyl group, reduction of the nitro and subsequent esterification of the carboxylic acid function leads to the desired product.

book
Clayden, Greeves, Warren and Wothers Organic Chemistry Oxford University Press 2001
Physico-Chemical Properties of Benzocaine¶
Some physico-chemical properties of benzocaine are listed below. The molecular weight, logP, the number of H-bond donors and H-bond acceptors can be compared to Lipinski's rule of five: all the values are within the framework of this rule.

Aspirin¶
Aspirin is a very popular analgesic agent with a relatively simple structure. An outline of the story associated with the discovery of aspirin can be found in a special chapter in this course. This drug can be synthesized as described in the following pages.

Aspirin Synthesis¶
Salicylic acid can be the starting material for aspirin since it is a readily available compound. A simple esterification step leads to the desired drug.

book
K.L. Williamson Macroscale and Microscale Organic Experiments D.C. Health and Company. 1989
Physico-Chemical Properties of Aspirin¶
Some physico-chemical properties of aspirin are listed below. The molecular weight, logP, the number of H-bond donors and H-bond acceptors can be compared to Lipinski's rule of five: all the values are within the framework of this rule.

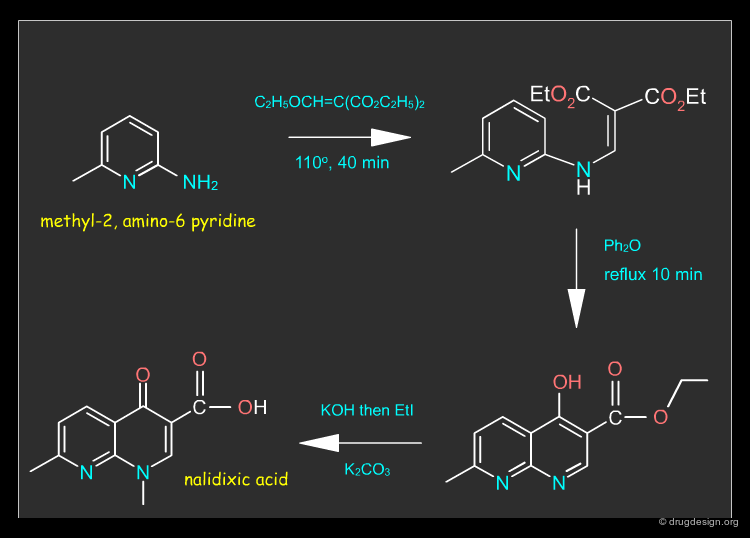
Nalidixic Acid¶
Nalidixic acid is a synthetic chemotherapeutic agent which works against gram-negative bacteria. The drug can be synthesized as described in the following pages.

Retrosynthetic Scheme¶
A possible retrosynthetic scheme for the synthesis of nalidixic acid is presented below; it is based on a substituted pyridine as starting material.

Nalidixic Acid Synthesis¶
Nalidixic acid can be synthesized in three steps from methyl-2, amino-6 pyridine, as described in the synthetic route below.
articles
8-Naphthyridine Derivatives. A New Class of Chemotherapeutic Agents George Y. Lesher, Ernest J. Froelich, Monte D. Gruett, John Hays Bailey, and R. Pauline Brundage J. Med. Chem. 5 1962
Physico-Chemical Properties of Nalidixic Acid¶
Some physico-chemical properties of nalidixic acid appear below. The molecular weight, logP, the number of H-bond donors and H-bond acceptors can be compared to Lipinski's rule of five: all the values are within the framework of this rule.

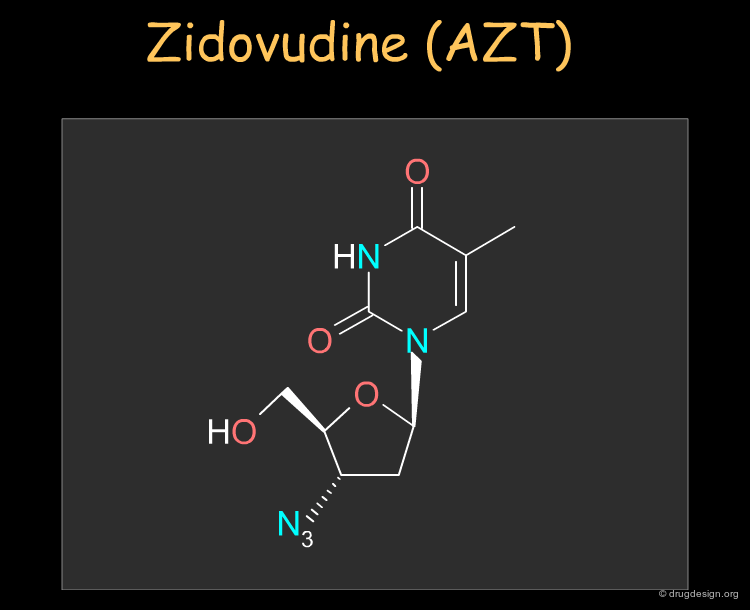
Zidovudine (AZT)¶
Zidovudine (AZT) was the first nucleoside reverse transcriptase inhibitor antiviral agent discovered and used to treat human immunodeficiency virus (HIV) that causes AIDS. The drug can be synthesized as described in the following pages.
Retrosynthetic Scheme¶
The retrosynthetic scheme shown below indicates that AZT 1 can be synthesized in four steps, starting from building block 5.

AZT Synthesis¶
In the first step the primary alcohol of (2'-deoxy-lyxo furanosyl) thymine 1 is protected selectively with trityl chloride. In the second step the secondary alcohol is activated, using MeSO2Cl and a nucleophilic displacement reaction. Treatment of the mesyl derivative with lithium azide leads to product 4. The trityl group is then deprotected under acidic conditions to give AZT.

articles
Nucleosides. V. The Monomesylates of 1-(2?-Deoxy-?-D-lyxofuranosyl)thymine Jerome P. Horwitz, Jonathan Chua, and Michael Noel J. Org. Chem. 29 1964
Nucleotide synthesis. IV. Phosphorylated 3'-amino-3'-deoxythymidine and 5'-amino-5'-deoxythymidine and derivatives Ronald P. Glinski, M. Sami Khan, Richard L. Kalamas, and Michael B. Sporn J. Org. Chem. 38(25) 1973
** ** C.K. Chu et al. Tetrahedron Letters 29 1988
Another Synthesis for AZT¶
AZT can be synthesized in different ways. Another example of synthesis is given below, which starts from D-mannitol. The key reactions in this scheme are Witting and Michael reactions. Move the cursor over the 'i' symbols to display the reaction conditions.

articles
Nucleosides. V. The Monomesylates of 1-(2?-Deoxy-?-D-lyxofuranosyl)thymine Jerome P. Horwitz, Jonathan Chua, and Michael Noel J. Org. Chem. 29 1964
Nucleotide synthesis. IV. Phosphorylated 3'-amino-3'-deoxythymidine and 5'-amino-5'-deoxythymidine and derivatives Ronald P. Glinski, M. Sami Khan, Richard L. Kalamas, and Michael B. Sporn J. Org. Chem. 38(25) 1973
** ** C.K. Chu et al Tetr. Lett. 29 1988
Which Route is Preferable?¶
The synthesis that starts from D-mannitol is more economic. Thymidine the starting material for the first scheme is very expensive whereas D-mannitol is readily available. Today not only economic factors but also ecologic factors are taken into account and chemists are paying more attention to green chemistry.

book
Anastas, P. T.; Warner, J. C. Green Chemistry: Theory and Practice Oxford University Press, New York 1998
Physico-Chemical Properties of Zidovudine¶
Some physico-chemical properties of Zidovudine are presented below. The molecular weight, logP, the number of H-bond donors and H-bond acceptors can be compared to Lipinski's rule of five: all the values are within the framework of this rule.

Terfenadine¶
Histamine is released in a variety of allergic conditions and mediates its activity by interaction with H1 receptors. Terfanidine is H1 blocker of the second generation and is used for the treatment of seasonal allergic rhinitis. The drug can be synthesized as described in the following pages.

Retrosynthetic Scheme¶
A possible retrosynthetic scheme for the synthesis of Terfenadine is indicated below. The synthesis is based on the substituted piperidine 3, and the chloro-ketone 4.
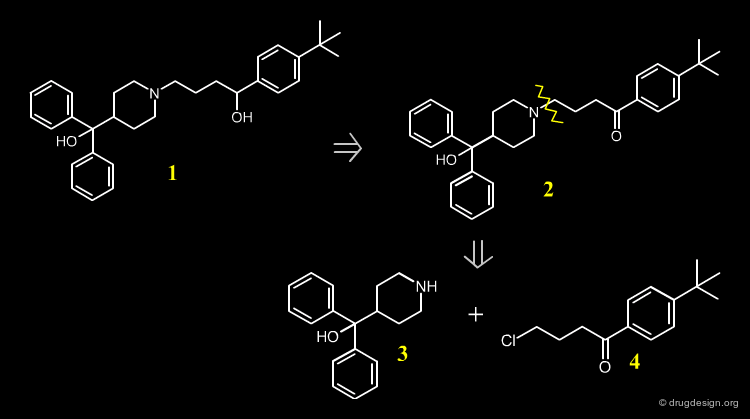
Terfenadine Synthesis¶
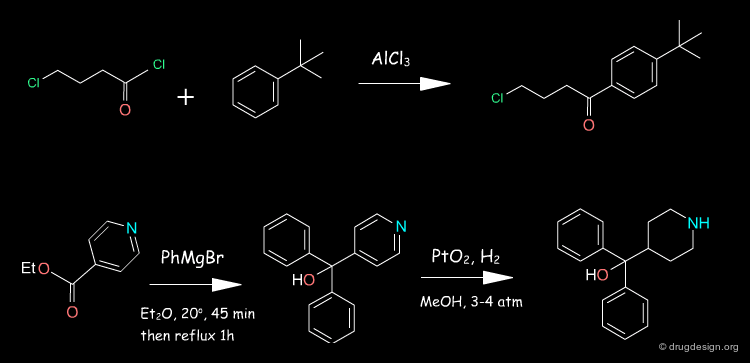
The 4-pyridine methanol is obtained using isonicotinic ethyl ester and a double Grignard reaction with phenyl magnesium bromide. Hydrogenation follows, in the presence of PtO2. The obtained azacyclonol is alkylated with the chlorobutanone in refluxing toluene in the presence of an inorganic base and a catalytic amount of NaI. Terfenadine is obtained using a reducing agent KBH4 as a racemic alcohol.

articles
Terfenadine Albert A. Carr et al Arzneim Forsch 32 1982
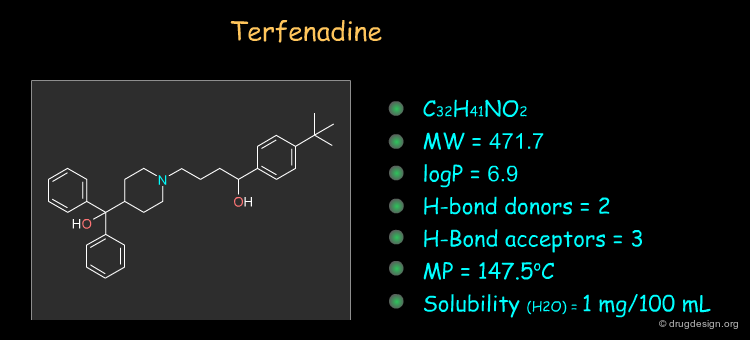
Physico-Chemical Properties of Terfenadine¶
Some physico-chemical properties of terfenadine are listed below. The molecular weight, logP, the number of H-bond donors and H-bond acceptors can be compared to Lipinski's rule of five: all the values are within the framework of this rule.
Nifedipine¶
Nifedipine was the first drug approved in the dihydropyridine series. It is a calcium channel blocker and it acts in the treatment of angina and hypertension. This drug can be synthesized as described in the following pages.

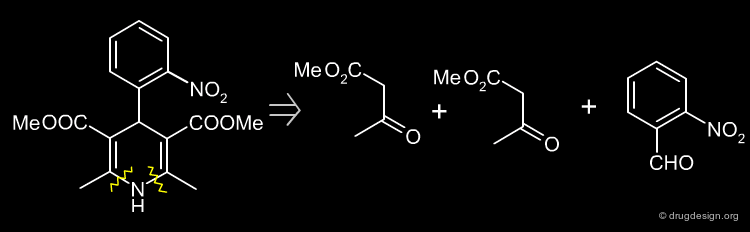
Simple Retrosynthesis of Nifedipine¶
The following scheme describes a simple retrosynthesis of Nifedipine starting from 2-nitrobenzaldehyde.
One-Pot Synthesis of Nifedipine¶
The corresponding synthesis of Nifedipine can be developed in a 'one-pot' reaction (Hantzsch condensation). However, it yields several products and therefore is not optimal.

Two Steps Retrosynthesis Scheme of Nifedipine¶
The retrosynthesis of Nifedipine presented here involves two steps, but is more preferable because it does not have the drawback of impurities.
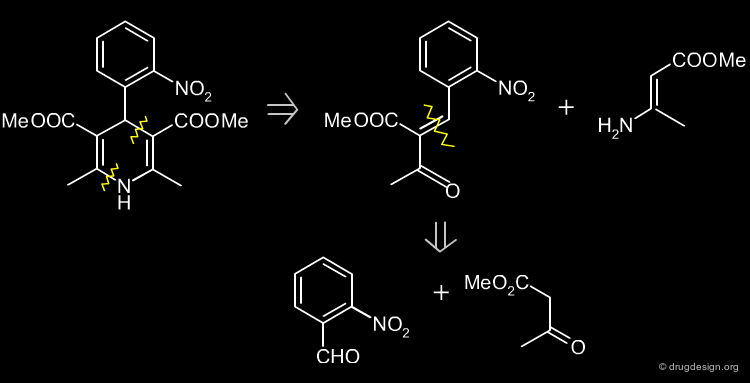
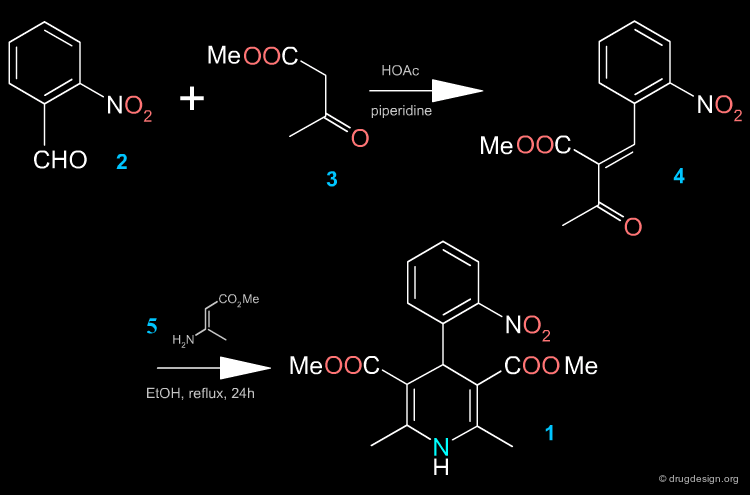
Two Steps Synthesis of Nifedipine¶
In the first step, the benzylidine ester 4 is produced (intermediate Knoevenagel product). A Hantsch reaction follows with the ethyl 3-aminocrotonate 5, which leads to the desired product.
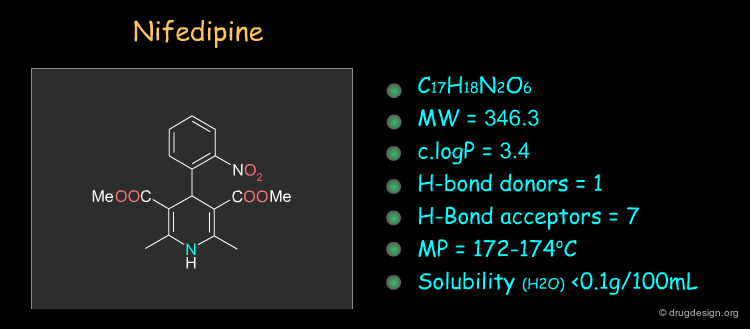
Physico-Chemical Properties of Nifedipine¶
Some physico-chemical properties of Nifedipine are listed below. The molecular weight, logP, the number of H-bond donors and H-bond acceptors can be compared to Lipinski's rule of five: all the values are within the framework of this rule.
Sildenafil (Viagra)¶
Sildenafil (Viagra) is a drug used to treat impotence in men. The molecule can be synthesized as described in the following pages.

Retrosynthetic Scheme¶
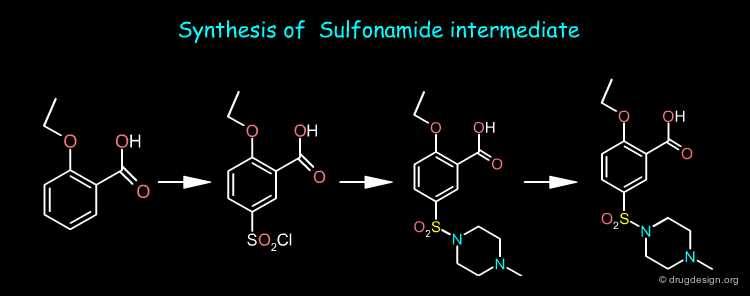
A retrosynthesis of Sildenafil involves only two precursors: the nitropyrazole and the sulfonamide intermediates shown below.

Sildenafil Synthesis¶
The nitropyrazole intermediate can be prepared from methylhydrazine, estermalonate and 2-pentanone as starting materials whereas the sulfonamide intermediate is prepared from o-ethoxy benzoic acid. Sildenafil is obtained by first reducing the nitro of the nitropyrazole and then coupling the two pieces and cyclization of the pyrimidone ring.
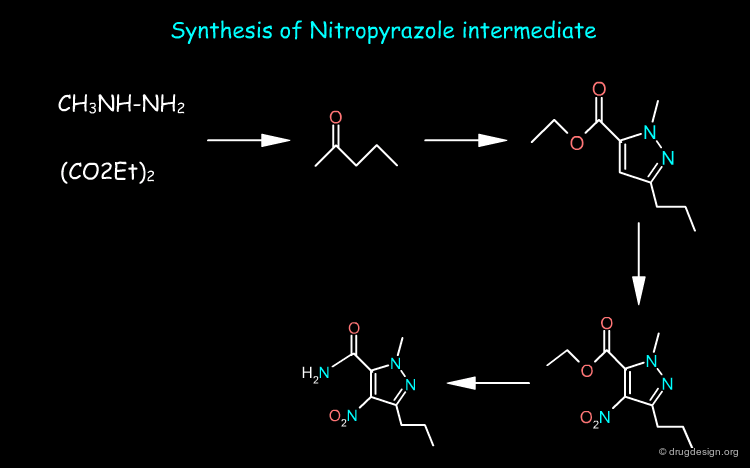
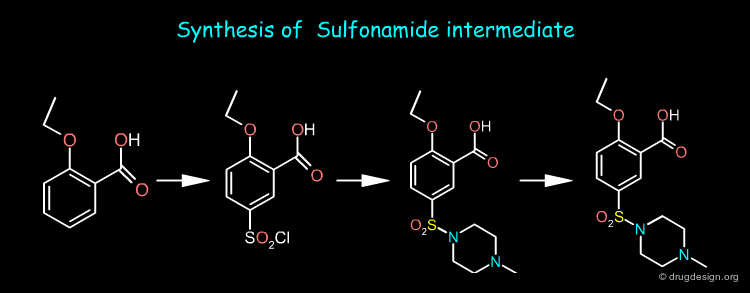

articles
The development of an environmentally benign synthesis of sildenafil citrate (Viagra ) and its assessment by Green Chemistry metrics Peter J. Dunn, Stephen Galvin and Kevin Hettenbach Green Chemistry 6(1) 2004
Physico-Chemical Properties of Sildenafil¶
Some physico-chemical properties of Sildenafil are listed below. The molecular weight, logP, the number of H-bond donors and H-bond acceptors can be compared to Lipinski's rule of five: all the values are within the framework of this rule.

Programs for Computer-Aided Synthesis¶
Programs for Computer-Aided Synthesis¶
Programs for computer-aided synthesis started to appear in the late 1960s. Important progress has been made; however system development was hampered by the reluctance of chemists to use such programs. Useful for the exploration of synthetic routes, these programs may be disappointing when utilized as black boxes to find a relevant route for the synthesis of a complex molecule. A brief outline of some of these systems is given in the following pages.

articles
Computer-assisted Synthesis N.S. Zefirov and E.V. Gordeeva Uspekhi Khimii 56(10) 1987
Cheminformatics and Organic Chemistry. Computer-Assisted Synthetic Analysis Martin Ott Cheminformatics (IOS Press) 1 Numbers 1-2 2004
Computer-aided organic synthesis Mattwew Todd Chem Soc Rev 34(3) 2005
book
R. Barone, M. Chanon Encyclopedia of Computational Chemistry John Wiley and Sons, Chichester 1998
Retrosynthetic Programs¶
When a chemical reaction is viewed "retrosynthetically" it is considered as going backwards from a target molecule to the starting materials. The main aim of retrosynthetic strategies is to generate precursors that correspond to available starting material. In the following pages examples of retrosynthetic programs are presented.

LHASA¶
LHASA (Logic and Heuristics Applied to Synthetic Analysis) was developed in the early 1960s in a group headed by E.J. Corey at Harvard, and was one of the first major systems made for computer-assisted synthetic design. It was initially developed to implement Corey's rules for synthetic chemistry (1967) where he defined novel concepts such as 'retrosynthesis', 'synthons' and 'disconnections'. Since its introduction LHASA has been actively maintained and enhanced. The system was and continues to be used by many groups worldwide.

articles
Algorithm for machine perception of synthetically significant rings in complex cyclic organic structures E. J. Corey and George A. Petersson J.Am.Chem.Soc. 94 1972
General methods of synthetic analysis. Strategic bond disconnections for bridged polycyclic structures E. J. Corey, W. Jeffrey Howe, H. W. Orf, David A. Pensak, and George Petersson J.Am.Chem.Soc. 97 1975
Computer-assisted synthetic analysis. Synthetic strategies based on appendages and the use of reconnective transforms E. J. Corey and William L. Jorgensen J.Am.Chem.Soc. 98 1976
SYNCHEM¶
SYNCHEM was developed in the early 1970s by H.L. Gelernter at Sunny Brook (NY) for automatic organic synthesis generation using artificial intelligence and heuristic computer programming. The method was based on a knowledge base containing data that were published on the chemical reactions involved and a small catalogue of readily available starting material. Challenged by synthetic experts the program was criticized for giving "naive" solutions too often. Attempts were made in 1978 to improve the first version.

articles
Empirical Explorations of SYNCHEM Gelernter, H.L., Sanders, A.F., Larsen, D.L., Agarwal, K.K, Boivie, R.H., Spritzer, G.A., and Searleman, J.T. Science 197 Sept. 9, 1977
A Computer-Oriented Linear Canonical Notational System for the Representation of Organic Structures with Stereochemistry. Krishna K. Agarwal, Herbert L. Gelernter Journal of Chemical Information and Computer Sciences 34(3) 1994
Building and refining a knowledge base for synthetic organic chemistry via the methodology of inductive and deductive machine learning Herbert L. Gelernter, J. Royce Rose, Chyouhwa Chen Journal of Chemical Information and Computer Sciences 30(4) 1990
Application of Chemical Transforms in SYNCHEM2 K. Agarwal, D. Larsen, and H. Gelernter Computers and Chemistry 2 1978
A Computer-Oriented Linear Canonical Notational System for the Representation of Organic Structures with Stereochemistry K. Agarwal and H. Gelernter Journ. Chem. Inf. Comp. Sci. 34 1994
SECS, OCCS, CASP¶
SECS (Simulation and Evaluation of Chemical Synthesis) was developed in 1978 by W.T. Wipke. The program recognizes stereochemical features. In addition 3D energy minimizations can be performed to assess whether a reactive site is accessible or subject to a particular steric hindrance before a suitable transform is proposed. The predecessor of SECS was OCCS (Organic Chemical Simulation of Synthesis - 1969). The CASP (Computer Aided Synthesis Planning System) system developed and used in the 1980s by a consortium of European pharmaceutical companies is an offshoot of the SECS program.

articles
Simulation and Evaluation of Chemical Synthesis - SECS: An Application of Artificial Intelligence Techniques W. Todd Wipke, Glenn I. Ouchi, S. Krishnan Artif. Intell. 11(1-2) 1978
Formal and Mathematically-Based Programs¶
Formal and mathematically based programs use theoretical descriptions of possible chemical reactions. The formalism can be based on matrices, a set of heuristics or symbolic representations of bond formation and cleavage. In the following pages some programs based on such formal descriptions of chemical reactions are described.

IGOR¶
IGOR (Interactive Generation of Organic Reactions) was developed in the late 1970s by I. Ugi as a logic-based synthetic analysis program. A molecule is represented as a matrix; chemical reactions are also represented by matrices. The product of a reaction is obtained by adding the corresponding reaction matrix to the starting material matrix. The mathematical framework provides a convenient way to describe and classify reactions. IGOR contributed to the discovery of new reactions, which were later shown to be possible experimentally. The program is interactive and driven by the user who provides chemical knowledge, intuition and preferences.

articles
Computer-Assisted Solution of Chemical Problems: A New Discipline in Chemistry Ivar Ugi, Johannes Bauer, Klemens Bley, Alf Dengler, Andreas Dietz, Eric Fontain, Bernhard Gruber, Rainer Herges, Michael Knauer, Klaus Reitsam, Natalie Stein Angew. Chem., Int. Ed. Engl. 32 1993
Computer-Assisted Solution of Chemical Problems: A New Discipline in Chemistry Ivar Ugi, Johannes Bauer, Klemens Bley, Alf Dengler, Andreas Dietz, Eric Fontain, Bernhard Gruber, Rainer Herges, Michael Knauer, Klaus Reitsam, Natalie Stein Angew. Chem., Int. Ed. Engl. 32 1993
EROS¶
EROS (Elaboration of Reactions for Organic Synthesis) was developed in the late 1970s by the group of J. Gasteiger in Germany. It was designed as a reaction generation program with the intention of limiting the combinatorial explosion of the synthetic tree. The acceptance or the rejection of reaction steps is based on thermodynamic considerations (reaction enthalpies, bond dissociation energies) and electronic parameters (atomic charges, inductive, resonance, polarizability effects). EROS contributed to the discovery of a simple industrial route to Acrylonitrile.

articles
EROS - A Computer Program for Generating Sequences of Reactions J. Gasteiger, C. Jochum Topics Curr. Chem. 74 1978
Computer-assisted Reaction Prediction and Synthesis Design J. Gasteiger, W.-D. Ihlenfeldt, P. Roese, R. Wanke Anal. Chim. Acta 235 1990
A New Treatment of Chemical Reactivity: Development of EROS, an Expert System for Reaction Prediction and Synthesis Design J. Gasteiger, M. G. Hutchings, B. Christoph, L. Gann, C. Hiller, P. Loew, M. Marsili, H. Saller, K. Yuki Topics Curr. Chem. 137 1987
SYNGEN¶
SYNGEN (SYNthesis GENeration) is a program introduced in the late 1970s by J.B. Hendrickson at Brandeis where he developed a novel approach for the generalization of organic functionality and reactions based on a symbolic representation of bond formation and cleavage. The program tries to find all the most efficient routes - the 'ideal synthesis' being the shortest and most atom-economical. SYNGEN is accompanied by another program enabling the interactive analysis of synthetic routes.

articles
Organic Synthesis in the Age of Computers James B. Hendrickson Angew. Chem., Int. Ed. Engl. 29, Issue 11 November 1990
A general protocol for systematic synthesis design James B. Hendrickson Top Curr Chem. 62 1976
A logic-based program for synthesis design James B. Hendrickson, David L. Grier, and A. Glenn Toczko J.Am.Chem.Soc. 107 1985
Comprehensive System for Classification and Nomenclature of Organic Reactions James B. Hendrickson J.Chem.Inf.Comput.Sci. 37 1997
RAIN¶
RAIN (Reactions And Intermediates Networks) is a reaction generator program that generates reactions and reaction schemes from reactions and structures by taking both the synthetic and retrosynthetic directions into account. RAIN was introduced by the Ugi group in 1987 using the same algebraic model of constitutional chemistry as in IGOR. The method is useful for elucidating reaction mechanisms.

articles
Computer Assisted Bilateral Generation of Reaction Networks from Educts and Products E. Fontain, J. Bauer, I. Ugi Chem. Lett.
1987
The Generation of Reaction Networks with RAIN. 1. The Reaction Generator E. Fontain, K. Reitsam J. Chem. Inf. Comput. Sci. 31 1991
The Generation of Reaction Networks with RAIN. 2. Resonance Structures and Tautomerism E. Fontain Tetrahedron Comput. Methodol. 3 1990
Forward Reaction Predictions¶
Forward reaction predictions correspond to approaches that develop in the synthetic direction; therefore they begin with the starting material. Examples of starting-material oriented search programs are presented in the following pages.

CAMEO¶
CAMEO (Computer-Assisted Mechanistic Evaluation of Organic Reactions) is a program developed in the early 1980s by W.I. Jorgensen for the prediction of the mechanism and products of an organic reaction. The program is instructed for the identification of electrophiles, nucleophiles and pKa values. Given the starting materials, reagents and conditions, the goal of the program is to predict all possible products that may be produced. CAMEO has been tested successfully against a number of published synthetic works.

articles
CAMEO: a program for the logical prediction of the products of organic reactions William L. Jorgensen, Ellen R. Laird, Alan J. Gushurst, Jan M. Fleischer, Scott A. Gothe, Harold E. Helson, Genevieve D. Paderes, and Shenna Sinclair Pure and Applied Chem. 62 (10) 1990
Computer-assisted mechanistic evaluation of organic reactions. 1. Overview Timothy D. Salatin and William L. Jorgensen J.Am.Chem.Soc. 45 1980
CHIRON¶
CHIRON was developed by S. Hanessian (1990) as a starting material-oriented program to reveal appropriate starting material. The program searches for maximal similarity in terms of carbon skeleton. Stereochemistry and functionality, is searched by the program between the target and databases of available chemicals. CHIRON has proven capable of finding starting material for complex targets with many chiral centers.

articles
The Psychobiological Basis of Heuristic Synthesis Planning -Man, Machine and the Chiron Approach S. Hanessian, J. Franco and B. Larouche Pure and Applied Chem. 62 1990
Computer-Assisted Analysis and Perception of Stereochemical Features in Organic Molecules Using the Chiron Program S. Hanessian, J. Franco, G. Gagnon, D. Larame and B. Larouche J. Comp. Chem. Inf. Sci. 30 1990
WODCA¶
WODCA (Workbench for the Organization of Data for Chemical Applications) was developed in 1995 by J. Gasteiger's group in Germany. This software incorporated the EROS reaction generation program into the broader WODCA range of programs. It can assist the chemist in starting-material search or for retrosynthetic analyses for either single structures or combinatorial libraries. The program automatically performs similarity searches in catalogues of available chemicals, defines strategic bonds and undertakes searches in reaction databases.

articles
Collection of Computer Methods for Synthesis Design and Reaction Prediction J. Gasteiger, W.-D. Ihlenfeldt, P. Roese Recl. Trav. Chim. Pays-Bas 111 1992
Similarity Concepts for the Planning of Organic Reactions and Syntheses J. Gasteiger, W.-D. Ihlenfeldt, R. Fick, J. R. Rose J. Chem. Inf. Comput. Sci. 32 1992
Computer-Assisted Design of Syntheses for Heterocyclic Compounds R. Fick, W.-D. Ihlenfeldt, J. Gasteiger Heterocycles 40 1995
Computer-Assisted Synthesis and Reaction Planning in Combinatorial Chemistry J. Gasteiger, M. Pfoertner, M. Sitzmann, R. Hoellering, O. Sacher, T. Kostka, N. Karg Persp. Drug Discov.Design 20 2000
Other Programs¶
Other programs for computer-aided synthetic planning are indicated in the following pages.

AIPHOS¶
AIPHOS (Artificial Intelligence for Planning and Handling Organic Syntheses) was developed in 1988 at Toyohashi in Japan by S.I. Sasaki and K. Funatsu for the automated development of synthetic routes. The program automatically analyses the target compound, selects suitable starting material and reduces strategic sites and their corresponding reaction paths.

articles
Computer-Assisted Organic Synthesis Design and Reaction Prediction System, 'AIPHOS' Funatsu, K.; Sasaki, S. I. Tetrahedron Comput. Methodol. 1 1988
Development of a Program for Construction of a Starting Material Library for AIPHOS, Koji Satoh, Shukou Azuma, Hiroko Satoh And Kimito Funatsu Journal of Chemical Software 4 1997
CAESA¶
CAESA (Computer Assisted Estimation of Synthetic Accessibility) is a computer program introduced in 1991 by G.J. Myatt at the University of Leeds to automatically rank sets of molecules according to their ease of synthesis and to display the synthetic routes. It also assesses the development of viable synthetic approaches by identifying suitable, readily available starting materials. The software uses expert system technology to mimic the methods used by an experienced synthetic chemist to estimate the ease of synthesis of a particular compound.

AOCR¶
AOCR (Automated Organic Chemical Reactions) is program developed by L. Ermanni (1993) based on a mathematical representation of organic synthesis, from the input of an organic compound. The program makes it possible to find all the syntheses of the target compound from the initial molecule given the reaction mechanisms. It also lists all the products of a reaction of the initial compounds.

SYSTEMATICHEM¶
SystematiChem is an organic chemistry software program for retrosynthesis and synthesis development. The program provides an automated synthesis pathway generation tool that allows the user to enter the compound and set a few minor parameters for the solution generation process. It then generates complete synthesis pathways using only known, purchasable starting materials and documented reactions. After this process is completed, the generated routes of the solutions can be reviewed and evaluated by the chemist using a solution viewer program.

Databases for Organic Synthesis¶
Databases for Organic Synthesis¶
A huge amount of printed information, data and databases have accumulated over the years in the area of organic synthesis and are regularly updated. This well documented and well-structured information constitutes an essential form of assistance for all synthetic chemists. Some of the most important sources of information are outlined in some detail in the following pages.

Printed Information¶
There is a vast number of textbooks, monograph series and articles that can be used by synthetic chemists as sources of information to find for example a detailed synthetic route, starting materials, conditions of a reaction, yields, catalysts, mechanisms etc... Examples of such printed information are shown below.


book
Methods of Organic Chemistry Thieme publisher 2001
Science of Synthesis: Houben-Weyl Methods of Molecular Transformations Georg Thieme Verlag, Stuttgart 2005
Organic Reactions Jossey-Bass publ. 61 volumes
Comprehensive Organic Chemistry Elsevier 1979
Comprehensive Organic Functional Group Transformations Elsevier 1995
Comprehensive Heterocyclic Chemistry I and II Pergamon Press 1984 - 1996
Comprehensive Natural Products Chemistry Pergamon Press 1999
Organic Syntheses
1921-2005
Encyclopedia of Reagents for Organic Syntheses Wiley 1995 +
Reagents for Organic Synthesis
1967 +
Handbook of Reagents for Organic Syntheses Wiley 2000
Comprehensive Organic Transformations Wiley 1999
The Chemistry of Functional Groups Wiley Guide 1992
Compendium of Organic Synthetic Methods Wiley
Advanced Organic Chemistry Jossey-Bass 2001
Synthetic Methods of Organic Chemistry Karger Publ.
Comprehensive Organic Synthesis Pergamon Press 1991 +
The Logic of Chemical Synthesis Wiley 1989
Protective Groups in Organic Synthesis Wiley 1999
Contemporary Organic Synthesis The Royal Society of Chemistry 1994 +
Specialized Abstracting Services¶
The Chemical Abstracts and Beilstein are two organizations that contributed substantially and for many years to the dissemination of scientific information in chemistry worldwide. Chemical Abstracts have existed for more than a century, whereas the Beilstein covers almost two centuries in chemistry. An outline of the resources provided by these two specialized abstracting services is given in the following pages.

Chemical Abstracts Services (CAS)¶
CAS was founded in 1907 with the aim of monitoring, abstracting, and indexing the world's chemistry-related literature. In total, CAS has indexed and summarized chemistry-related articles from more than 40,000 scientific journals representing about 24 million documents and more than 15 million substances that are accessible online through CAS. The data produced by CAS are accessible to research scientists in different formats, including the printed CA, CA on CD, the STN international online network, the SciFinder and SciFinder-Scholar desktop research tools. CAS is a division of the American Chemical Society.

SciFinder¶
SciFinder is used to access the CAS chemical information from a desktop with many types of searches. For reaction searches for example, once a structure is drawn, it is possible to refine the retrieved records with constraints such as the yield, the number of steps, the type of reaction etc...

Beilstein¶
The Beilstein database contains fully searchable chemical structures and reactions, associated chemical and physical properties, and detailed pharmacological and ecological data. The chronological index began in 1771 and addresses three primary types of data: substances, reactions and citations. The Beilstein database is updated quarterly by Elsevier, a subsidiary of MDL Information Systems Inc.

Gmelin¶
The Gmelin Database is the sister database to Beilstein, and has covered inorganic and organometallic compounds since 1772 to the present. It is used for graphic reaction searching. Gmelin is a product of MDL Information Systems.

Reaction Databases¶
There are a number of databases that are dedicated to chemical reactions or organic syntheses and provide useful practical information from the point of view of the synthesis. Some examples of such specialized databases (free or commercial) are indicated here with their associated (clickable) internet address.


On-Line Resources¶
If you are perplexed, have forgotten the content of a reaction referenced with a name (e.g. Stobbe condensation or Sakurai reaction) the on-line resources such as the "Organic Reaction Names" or the "Named Reactions in Organic Chemistry" might help you to rapidly recall it and view some examples. A vast number of resources are available on-line for the synthetic chemists; the list indicated below indicates only some of these. Portals such as the American Chemical Society or Organic Chemistry Worldwide Resources provide additional useful links.

Patent Databases¶
In the development phase of a drug, patent databases provide useful information on its synthesis which has not been published in the scientific articles or poster forms. Moreover, when engaged in a discovery project, it is important to know what types of structures are well covered by the competition. In the following pages some important patent databases are presented.

US Patents: USPTO¶
The United States Patent and Trademark Office (USPTO) provides free web access to its database of issued patents and published applications. Quick and advanced searches are possible with fields such as words, inventor name, inventor country, assignee name and country, issue date, examiner, patent number etc... The USPTO is an agency of the US Department of Commerce and the database is updated weekly.
European Patents: EPOLINE¶
The European Patent Register On-Line (EPOLINE) provides information on all European and Euro-PCT patent applications. Records include bibliographic data, legal status information but no description of the invention (claims or abstracts). Files are updated daily and information is available to online users 3-5 days after the action date.

Other Patent Databases¶
Other patent databases exist, some of which are indicated below. In particular, the IBM's Patent Server is a public service providing a different patent database from the US Patent abstracts with as service similar to that of the USPTO. Also, the Japanese Patent Office has a searchable database of Japanese patent abstracts, which include the patent number, title, inventor, company, and abstract of the patent.

Copyright © 2024 drugdesign.org