Success Stories in Drug Discovery¶
Info
Stories of the successful discovery of a selection of drugs are recounted in this chapter. Each story outlines the starting point for the project, the complete history of the discovery, barriers that had to be overcome, accidental observations, R&D breakthroughs, important dates, factors that contributed to the success and events that occurred following the discovery.
Number of Pages: 68 (±1 hours read)
Last Modified: June 2008
Prerequisites: None
Success Story-1 : Captopril¶
Captopril¶
Captopril was the first angiotensin-converting enzyme (ACE) inhibitor to be marketed (1981). The therapeutic concept, which aimed at reducing blood pressure by blocking the formation of the potent pressor agent angiotensin II from angiotensin I, through the inhibition of the Angiotensin Converting Enzyme, proved to be successful and was the foundation of an entirely new class of orally active antihypertensive drugs: the ACE inhibitors. The Captopril story is outlined in the following pages.

wikipedia
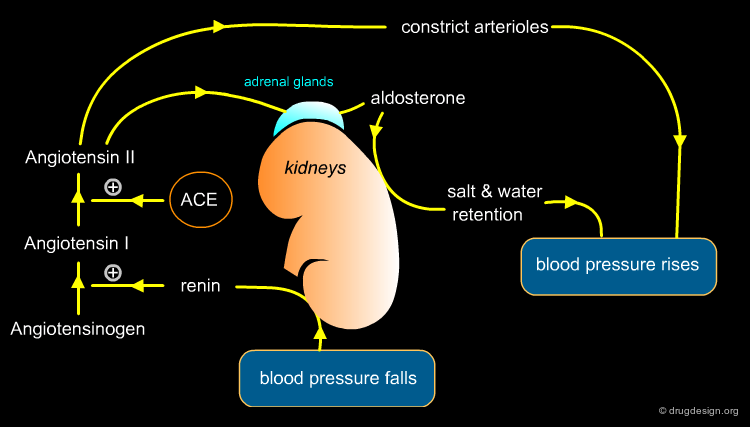
Captopril Target - ACE¶
The angiotensin converting enzyme (ACE) is the exopeptidase that catalyzes the production of angiotensin II through removal of the terminal dipeptide of angiotensin I. Angiotensin II causes potent vasoconstriction, a release of aldosterone and an increase in sodium absorption, all of which contribute to elevated blood pressure. Inhibiting ACE reduces production of Angiotensin II is therefore an ideal means of treating high blood pressure.

wikipedia
Starting Point: Venom Causes Drop in Blood Pressure¶
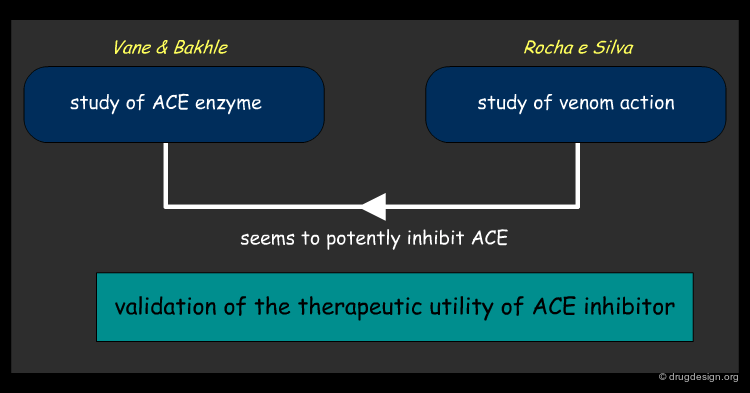
Intrigued by the drastic drop in blood pressure observed in a person bitten by a Brazilian viper, a team led by Rocha e Silva extracted the viper's venom to study its properties. One of Rocha e Silva's postdoctoral students went to London with samples of venom in the mid 1960s to work in the laboratory of Sir John Vane, who at that time was actively involved in the study of angiotensin I, angiotensin II and ACE.

Snake Venom Acts on the ACE Cascade¶
There were great reservations at that time about the role of Angiotensin I and II in hypertension but while studying the effects of the venom on the angiotensin system, Vane and Bakhle showed in 1968 that something in the venom appeared to be a potent inhibitor of ACE. Realizing that this discovery could be of importance in treating cardiovascular diseases, Vane, who was a consultant to the U.S. pharmaceutical company Squibb, suggested that they study the effects of the snake venom in order to find out whether angiotensin II was involved in regulating blood pressure.
The Captopril Story¶

articles
History of the design of captopril and related inhibitors of angiotensin converting enzyme D.W. Cushman and M.A. Ondetti Hypertension 17 1991
The Discovery of Captopril SMITH CG and VANE JR FASEB J 17(8) 2003
Developing an Assay for ACE¶
Despite great reluctance on the part of the Squibb research management, Cushman and Ondetti, an enzymologist and a synthetic peptide chemist, were finally put to work. Cushman began working at the Squibb institute in 1968 and had formulated the first quantitative assay for ACE. Cushman and Ondetti hypothesized that ACE was a zinc metalloprotease with a zinc ion at the heart of its enzymatic protein-cleavage action, based on early studies that had shown its inhibition using chelating agents such as EDTA and its reactivation using various divalent metal ions.

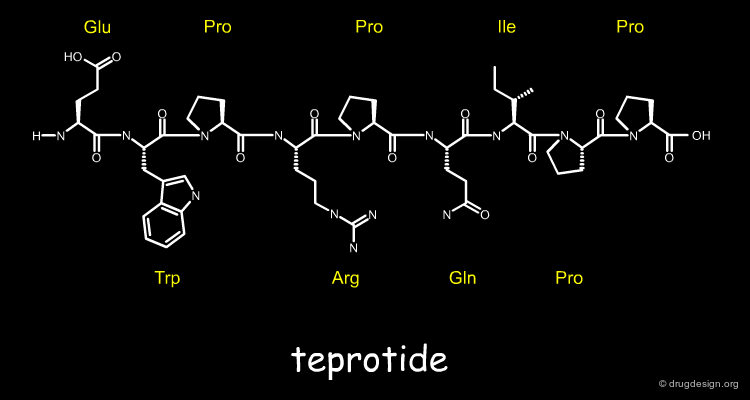
Isolating and Purifying the Venom Peptides¶
In 1970, they successfully isolated and purified six venom peptides that caused ACE inhibition, amongst them the peptide with the best duration of action, which was later called teprotide. They subsequently determined its chemical structure, a nona-peptide (9 amino acids) shown below.
articles
Inhibition of angiotensin-converting enzyme by analogs of peptides from Bothrops jararaca venom D. W. Cushman et al. Cellular and Molecular Life Sciences 29(8) 1973 10.1007/BF01930447
Encouraging Clinical Trial Results¶
A synthetic form of the teprotide entered into clinical trials where it was intravenously administered to patients. The first results were very encouraging as the compound had a blood pressure-lowering effect and confirmed the therapeutic validity of the project: ACE inhibitors could potentially be used as antihypertensive drugs.

articles
Estimating renin participation in hypertension: superiority of converting enzyme inhibitor over saralasin. Case DB, Wallace JM, Keim HJ, Weber MA, Drayer JI, White RP, Sealey JE, Laragh JH The American Journal of Medicine 61(5) 1976 10.1016/0002-9343(76)90160-1
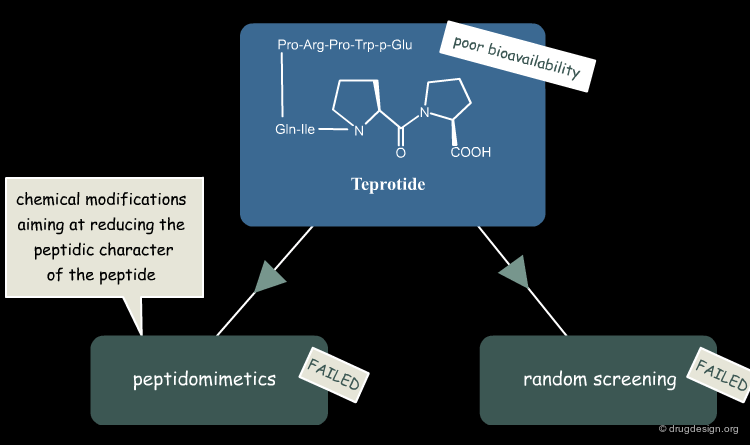
Project Virtually Abandoned at Squibb¶
Despite the promising blood pressure-lowering capability of teprotide, the peptide had low oral bioavailability due to its large size, which meant that it was unsuitable for development as a drug for chronic hypertension. Further peptidomimetic efforts and random screening of thousands of compounds did not yield a better candidate. The ACE-inhibitor project at Squibb was thus halted in 1972.
wikipedia
Back to the Project¶
In March 1974, Miguel Ondetti and David Cushman came across a publication by Byers and Wolfenden that detailed a new potent inhibitor of Carboxypeptidase-A (CPA): benzylsuccinic acid. They were very excited by this paper because CPA was a zinc-metalloprotease, as they had considered ACE to be. The new inhibitor of CPA combined in one molecule an analog of the terminal amino acid cleaved off during hydrolysis of the substrate and the liberated terminal carboxyl group of the other product. It was referred to as a bi-product inhibitor.
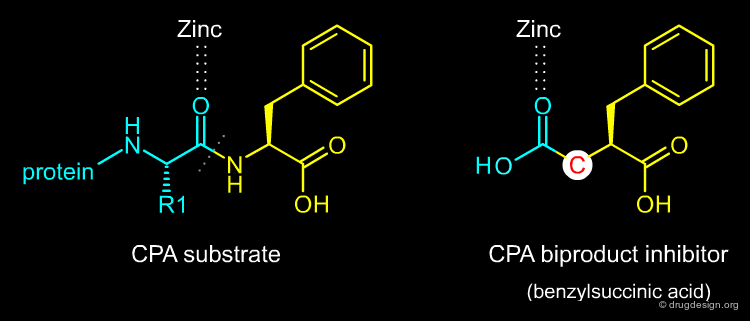
articles
Binding of the Bi-Product Analog Benzylsuccinic Acid by Carboxypeptidase A. Byers, L.D. Wolfenden, R. Biochemistry 12 1973 10.1021/bi00735a008
wikipedia
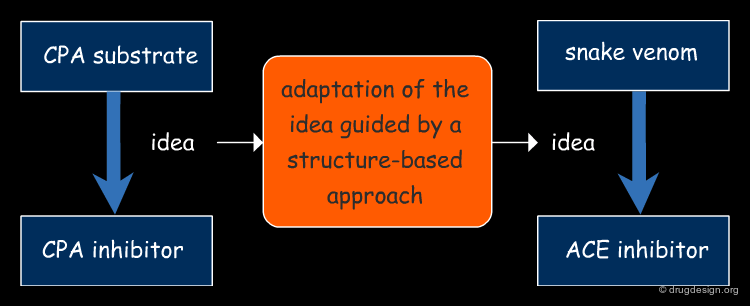
Applying the Concepts to ACE¶
Cushman and Ondetti returned to the ACE inhibitor project. This time around, rather than focusing on the structure of potential inhibitors, they decided to concentrate more on the structure of the active site of ACE based on its assumed similarity to CPA. Rather they imagined a new structure that in one molecule brought together some important elements present in both the snake venom and in the recently published CPA inhibitor.
The Basis of ACE and CPA Similarity¶
The basis for the assumption that ACE was like CPA was that they were both: (1) inhibited by metal chelating agents, (2) reactivated by manganese, zinc and cobalt, and (3) unable to cleave substrate analogs that possessed a terminal carboxamide-dicarboxylic amino acid.

X-ray Structure of CPA¶
Despite the small number of three-dimensional protein structures that had been determined by X-ray crystallography at that time, the structure of CPA had fortunately been discovered by Quiocho and Lipscomb in 1967. The knowledge of this structure was crucial to the development of a conceptual model of ACE's active site by Cushman and Ondetti.


articles
The structure of carboxypeptidase A. VII. The 2.0-angstrom resolution studies of the enzyme and of its complex with glycyltyrosine, and mechanistic deductions. Lipscomb WN, Hartsuck JA, Reeke GN Jr, Quiocho FA, Bethge PH, Ludwig ML, Steitz TA, Muirhead H, Coppola JC Brookhaven Symp Biol. 21(1) 1968
The structure of carboxypeptidase A. V. Studies of enzyme-substrate and enzyme-inhibitor complexes at 6 A resolution Steitz, T.A., Ludwig, M.L., Quiocho, F.A. and Lipscomb, W.N. J. Biol. Chem 242 1967
Modeling the Active Site of ACE (1/4)¶
Cushman and Ondetti constructed a conceptual model of the active site of ACE based on comparison with CPA. They concluded that the proposed active site of ACE had to possess the following characteristics: (1) A positively charged residue at the active site (analogous to Arg-145 of carboxypeptidase-A) which should bind with the negatively charged carboxyl group at the C-terminal of a peptide substrate.
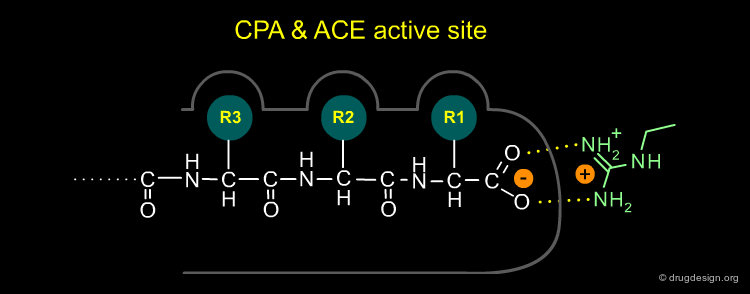
Modeling the Active Site of ACE (2/4)¶
(2) The zinc-ion of ACE, which is responsible for cleavage of the terminal dipeptide of angiotensin I, had to be bound to the enzyme at a place which is a dipeptide-length away from the aforementioned positively charged residue, unlike that of CPA which is a single amino-acid-length away

Modeling the Active Site of ACE (3/4)¶
(3) ACE, unlike CPA, showed no specificity for C-terminal hydrophobic amino acids and was therefore not expected to have a hydrophobic binding pocket as CPA does.

Modeling the Active Site of ACE (4/4)¶
(4) The ACE active site was presumed to contain a hydrogen-bond donor residue between the R1 and R2 binding pockets that would bind to the terminal, non-scissile, peptide bond of the substrate.
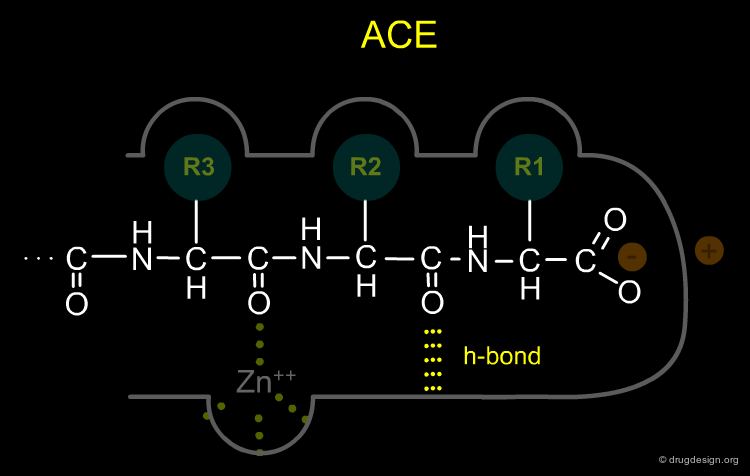
Design of a Novel ACE Inhibitor¶
Using the information they had gathered about the new CPA inhibitor and the characteristics of the active site, they surmised that the succinyl carboxyl group of the CPA inhibitor, benzyl succinic acid, would most likely interact with the central catalytic zinc ion of CPA. They further hypothesized that a succinylamino acid derivative, whose structure was similar to the dipeptide product of ACE action and had an additional zinc-binding succinyl carboxyl group, might inhibit ACE specifically.

The Phe-Ala-Pro Pharmacophore¶
At the first instance, this led them to succinyl proline, whose succinyl carboxyl group, they thought, would interact with the catalytic zinc ion. When tested, this compound did not have particularly good inhibitory properties but encouragingly, did have specificity for ACE. At the time that teprotide had been discovered, Cushman and Ondetti had carried out structure-activity studies on numerous venom peptides and their synthetic analogs; it emerged that a number of compounds that competitively inhibited ACE possessed the C-terminal amino acid sequence "Phe-Ala-Pro".

Finding a Lead Compound¶
Exploiting this optimal inhibitory "Phe-Ala-Pro" pharmacophore, Cushman and Ondetti subsequently synthesized the compound 2-methyl succinyl proline, which mimicked the terminal dipeptide Ala-Pro and still possessed a zinc-binding carboxyl group. This displayed better inhibition of ACE than succinyl proline and became the new lead compound.

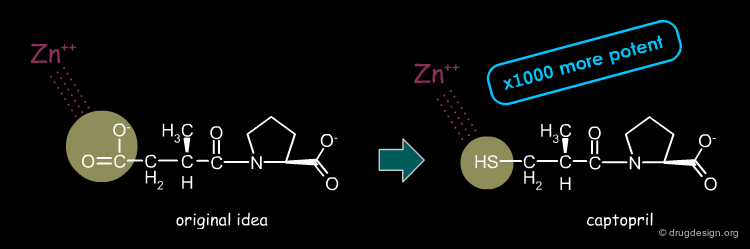
The Discovery of Captopril¶
During the subsequent optimization of this lead more than 60 different analogs were synthesized and tested by Cushman and Ondetti for their inhibition of ACE. They finally discovered the potent ACE inhibitor shown below, later named Captopril. The replacement of the carboxylate (expected to interact with the zinc atom) with a thiol group proved to be 1000 times more potent in inhibiting the enzyme than 2-methyl succinyl proline and 10-20 times more potent than the venom peptide, teprotide.
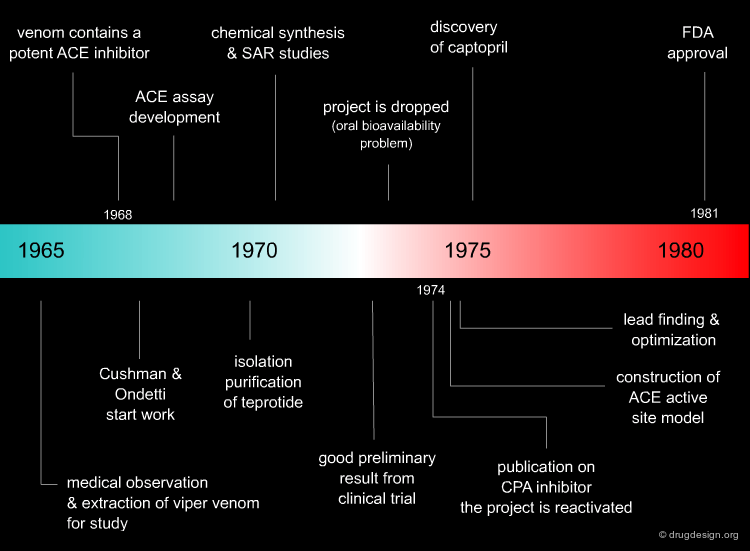
The Captopril Project Timeline¶
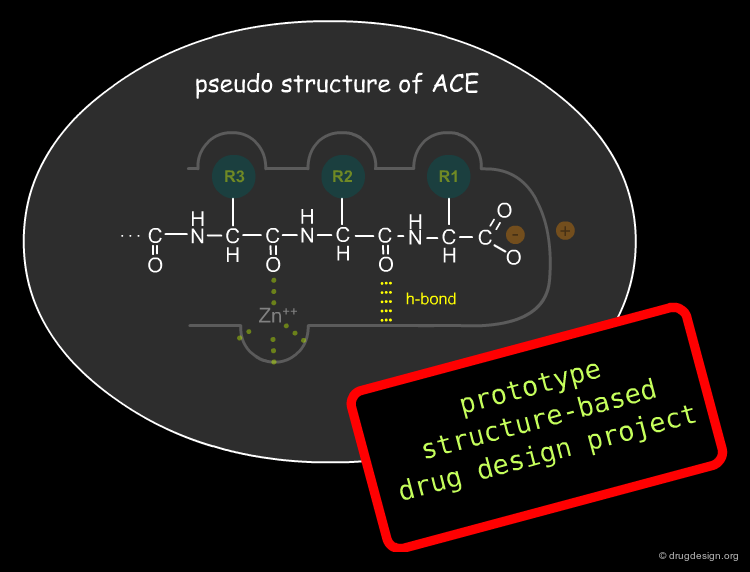
What Made the Success of the Project Possible?¶
If we try to retrospectively understand how this project turned into a success story despite the management's decision to stop it, aside from the persistence of the authors to continue searching, the major factors that contributed to the discovery of Captopril were the effective use of structure-based and ligand-based design approaches, as explained in the following pages. Note that serendipity also played a small part.

Structure-Based Component¶
Positive results were only achieved once Cushman and Ondetti changed their focus from the structure of known inhibitors to trying to understand the nature of the active site of ACE and its biological activity in terms of geometrical binding to the enzyme. The Captopril story is considered to be the first structure-based drug design project; their hypothetical model of the active site of angiotensin-converting enzyme was used to guide the design of potential small molecule inhibitors.
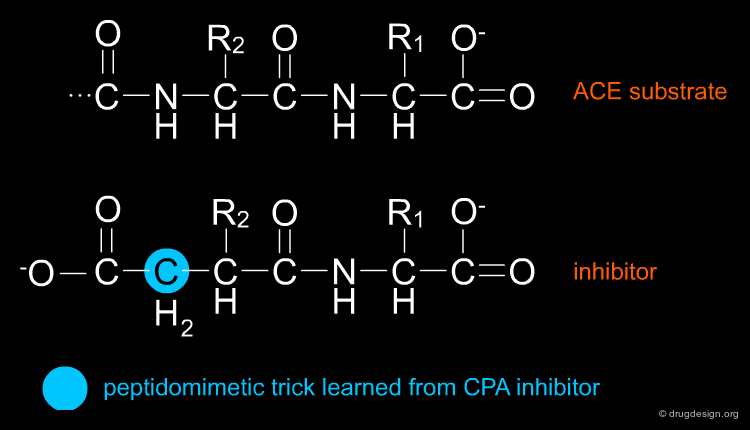
Ligand-Based Component¶
The structure activity studies that were carried out on the venom peptides and their synthetic analogs led to the identification of the pharmacophoric element (Phe-Ala-Pro). More importantly, the discovery of Benzylsuccinic acid, the Carboxypeptidase-A inhibitor described in the 1974 paper, was proven to be a crucial step in enabling the Squibb team to obtain an orally available drug.

Following the Discovery¶

Recent Structure of Captopril-ACE Complex¶
Although conceived from a structure-based perspective, the first X-ray complexes of inhibitors of ACE appeared only recently. The thiol group was found to bind to a Zinc atom and the carboxyl group forms hydrogen bonds with three residues: Lys-551, Tyr-520 and Gln-281 (and not arginine, as imagined 30 years earlier). It is interesting to note that the study reveals that the ACE protein actually bears little 3D similarity to that of carboxypeptidase-A.


Other Drugs in This Class¶
The design of Captopril enabled other companies to develop further ACE inhibitors for treatment of hypertension, some of them are shown below.

Success Story-2 : Aliskiren¶
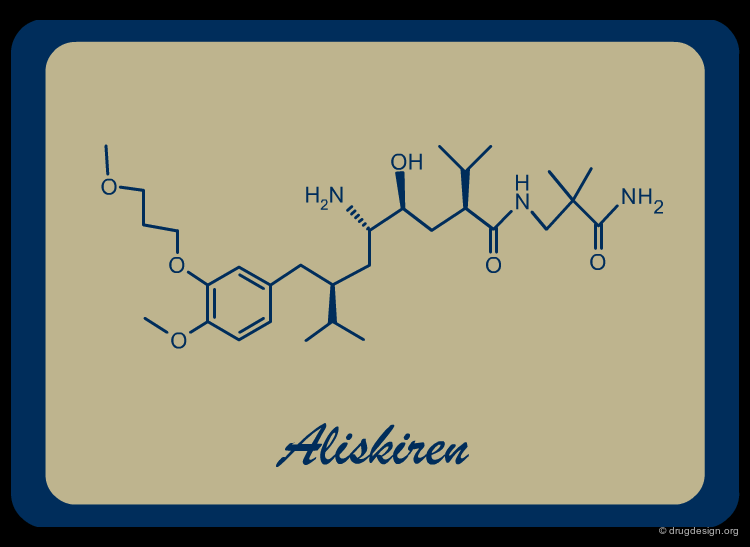
Aliskiren¶
Aliskiren (Tekturna, Rasilez) is the prototype of an entirely new class of antihypertensive drugs acting through the inhibition of the enzyme Renin and was introduced to the market in 2007. The Aliskiren story is presented in the following pages.
articles
Novel 2,7-Dialkyl Substituted 5(S)-Amino-4(S)-Hydroxy-8-Phenyl-Octanecarboxamide Transition State Peptidomimetics are Potent and Orally Active Inhibitors of Human Renin Richard Goeschke, Vittorio Rasetti, Nissim-Claude Cohen, Joseph Rahuel, Hans-Peter Baum, Peter Forgiarini, Christian R. Schnell, Markus G. Gruetter, Stefan Stutz, Grety Rihs, Trixie Wagner, Walter Fuhrer, Frederic Cumin, Jeanette M. Wood, Juergen Maibaum. J Med Chem 50 (20) 2007 10.1021/jm070314y
Structural Modification of the P2' Position of 2,7-Dialkyl Substituted 5(S)-Amino-4(S)-Hydroxy-8-Phenyl-Octanecarboxamides: The Discovery of Aliskiren, A Potent Non-Peptide Human Renin Inhibitor Active After Once Daily Dosing in Marmosets Juergen Maibaum, Stefan Stutz, Richard Goeschke, Pascal Rigollier, Yasuchika Yamaguchi, Frederic Cumin, Joseph Rahuel, Hans-Peter Baum, Nissim-Claude Cohen, Christian R. Schnell, Walter Fuhrer, Markus G. Gruetter, Walter Schilling, Jeanette M. Wood J Med Chem 50 (20) 2007 10.1021/jm070316i
Structure-based drug design and the discovery of aliskiren (Tekturna): perseverance and creativity to overcome a R and D pipeline challenge Nissim Claude Cohen Chem Biol Drug Des 70(6) 2007 10.1111/j.1747-0285.2007.00599.x
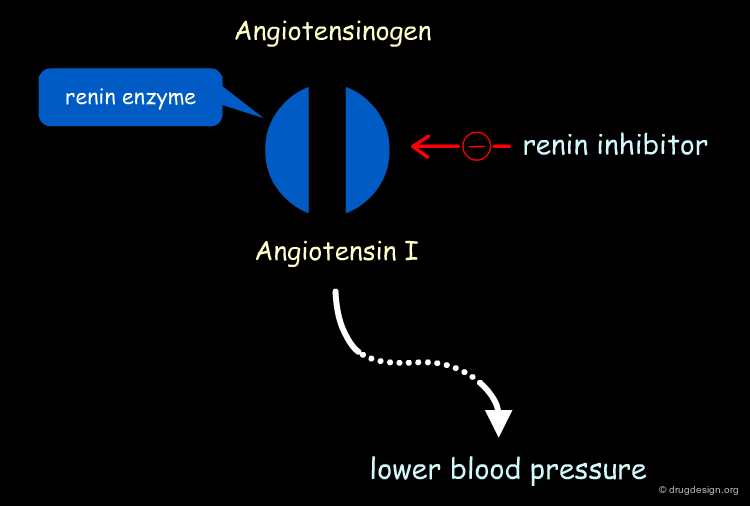
Aliskiren Target - Renin¶
Renin is secreted from the kidneys in response to a reduction in renal filtration pressure. The enzyme is found at the start of the Renin-Angiotensin cascade and is responsible for hydrolyzing Angiotensinogen, produced by the liver, to Angiotensin I. Angiotensin I is subsequently converted into Angiotensin II, which causes potent vasoconstriction, a release of aldosterone and an increase in sodium absorption, all of which contribute to elevated blood pressure. Renin inhibition was therefore thought to be an excellent means of reducing hypertension.

wikipedia
Starting Point¶
By the early 1980's the therapeutic concept of interrupting the renin-angiotensin cascade to lower blood pressure was well established, as evidenced by the successful commercialization of the ACE inhibitors (e.g. Captopril and related drugs). Although the ACE inhibitors were successful drugs, they possessed undesirable side effects (e.g. cough) that prompted pharmaceutical companies to search for new, better tolerated antihypertensive agents. Renin inhibition and Angiotensin-II (A-II) blockade were the two major R&D areas that were pursued.

articles
Cough associated with the use of captopril Sesoko S, Kaneko Y. Arch Intern Med 145(8) 1985
Cough associated with captopril and enalapril D M COULTER, I R EDWARDS British Medical Journal 294 1987
The Aliskiren Story¶

articles
Structure-based drug design and the discovery of aliskiren (Tekturna): perseverance and creativity to overcome a R and D pipeline challenge Nissim Claude Cohen Chem Biol Drug Des 70(6) 2007 10.1111/j.1747-0285.2007.00599.x
Novel 2,7-Dialkyl Substituted 5(S)-Amino-4(S)-Hydroxy-8-Phenyl-Octanecarboxamide Transition State Peptidomimetics are Potent and Orally Active Inhibitors of Human Renin Richard Goeschke, Vittorio Rasetti, Nissim-Claude Cohen, Joseph Rahuel, Hans-Peter Baum, Peter Forgiarini, Christian R. Schnell, Markus G. Gruetter, Stefan Stutz, Grety Rihs, Trixie Wagner, Walter Fuhrer, Frederic Cumin, Jeanette M. Wood, Juergen Maibaum. J Med Chem 50 (20) 2007 10.1021/jm070314y
Structural Modification of the P2' Position of 2,7-Dialkyl Substituted 5(S)-Amino-4(S)-Hydroxy-8-Phenyl-Octanecarboxamides: The Discovery of Aliskiren, A Potent Non-Peptide Human Renin Inhibitor Active After Once Daily Dosing in Marmosets Juergen Maibaum, Stefan Stutz, Richard Goeschke, Pascal Rigollier, Yasuchika Yamaguchi, Frederic Cumin, Joseph Rahuel, Hans-Peter Baum, Nissim-Claude Cohen, Christian R. Schnell, Walter Fuhrer, Markus G. Gruetter, Walter Schilling, Jeanette M. Wood J Med Chem 50 (20) 2007 10.1021/jm070316i
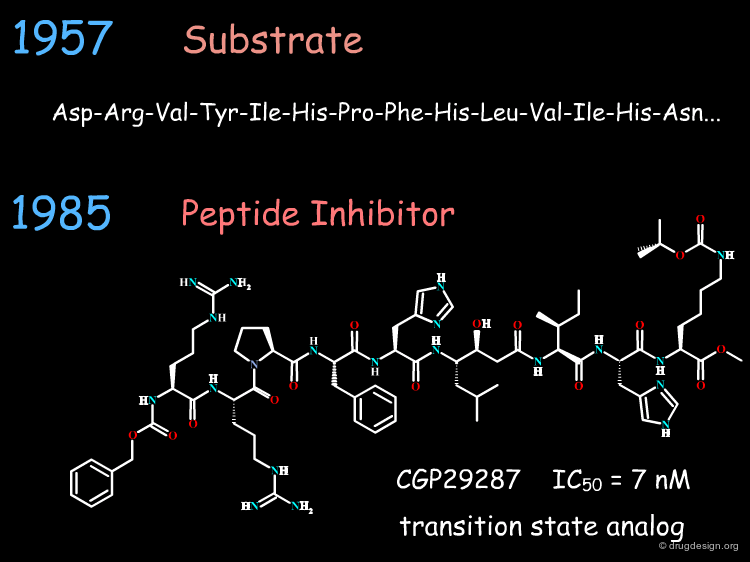
The First Generation of Renin Inhibitors¶
In the 1980's, pharmaceutical companies began to look at ways to design Renin inhibitors. The designs were based on the transition state analog approach. The figure below shows one example of a so-called 'transition state' inhibitor discovered in 1985 by Novartis (Ciba-Geigy); however, such a molecule could not be further developed because of its high molecular weight (1500 Daltons).
articles
Effects of a specific and long-acting renin inhibitor in the marmoset Wood JM, Gulati N, Forgiarini P, Fuhrer W, Hofbauer KG. Hypertension 7(5) 1985
wikipedia
The Second Generation of Renin Inhibitors¶
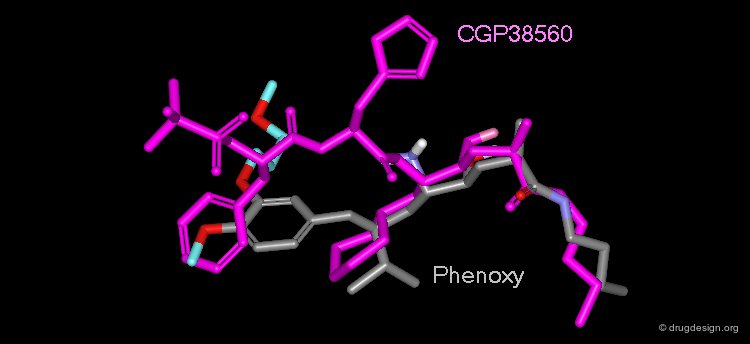
Development of the second generation of Renin inhibitors focused on diminishing both the molecular weight and the peptidic character of the first-generation inhibitors. CGP38560 is an example of such a molecule discovered at Novartis in 1988. However, clinical trials revealed there was low oral absorption and rapid biliary excretion of the molecule. The development of this drug candidate was thus dropped.

articles
CGP 38 560: orally active, low-molecular-weight renin inhibitor with high potency and specificity Wood JM, Criscione L, de Gasparo M, Buhlmayer P, Rueger H, Stanton JL, Jupp RA, Kay J. J Cardiovasc Pharmacol. 14(2) 1989
Peptidomimetic Approach was Unsuccessful¶
In fact many pharmaceutical companies tried this same approach and gave up for the same reasons. Here are some examples of drug candidates that were all abandoned.

The Need for a New Non-Peptidic Scaffold¶
The researchers at Novartis saw that making changes to the 2D peptide molecule in a step-wise manner in an attempt to reduce its peptidic character would not yield a successful candidate. They knew that they needed a completely different, non peptidic molecule. Subsequently, they tried the random screening approach, but this did not yield any hit that could serve as a basis for a synthetic program.

Novartis's New Rational Approach¶
The management at Novartis realized that the key to success was to look at the molecules in 3D and incorporate a modeling strategy into the project. In 1988, one modeler, Cohen and three medicinal chemistry teams led by Goeschke, Rasetti and Maibaum, began to tackle the issue by deploying a rational drug design approach.

3D Model of the Enzyme¶
As no X-ray structures of renin were available, one the team's first priorities was to obtain a 3D model of the target. The team used a homology model of renin developed by Blundell et al that was based on penicillopepsin. This model played an important role in the subsequent discovery of Aliskiren.

articles
Three-dimensional structure, specificity and catalytic mechanism of renin Blundell T, Sibanda BL, Pearl L. Nature 304(5923) 1983 10.1038/304273a0
Computer graphics modelling of human renin. Specificity, catalytic activity and intron-exon junctions Sibanda BL, Blundell T, Hobart PM, Fogliano M, Bindra JS, Dominy BW, Chirgwin JM. FEBS Lett. 174(1) 1984 10.1016/0014-5793(84)81086-8
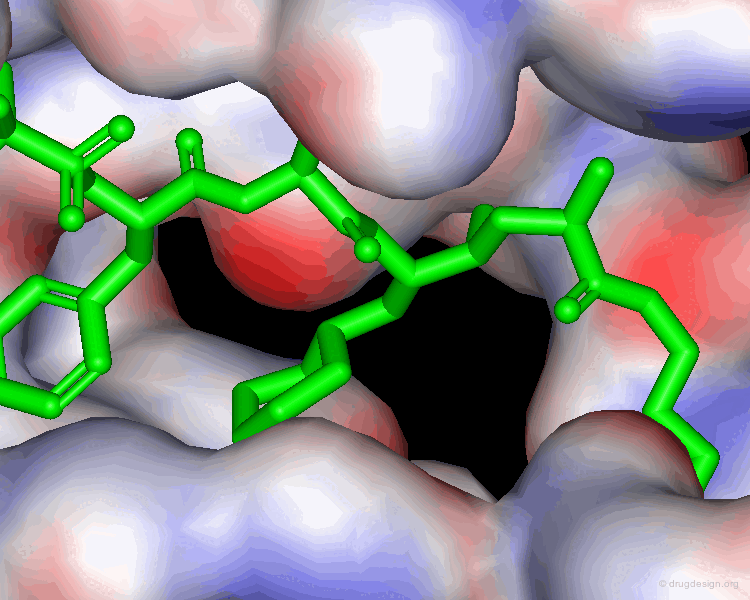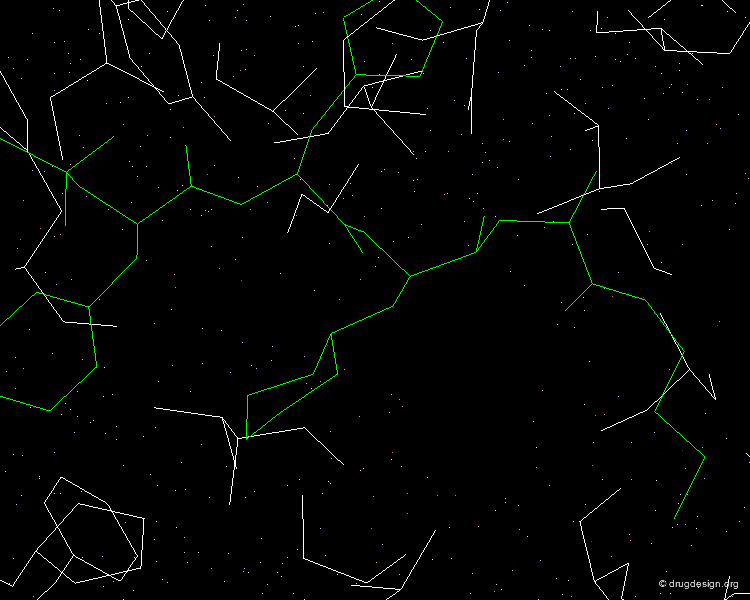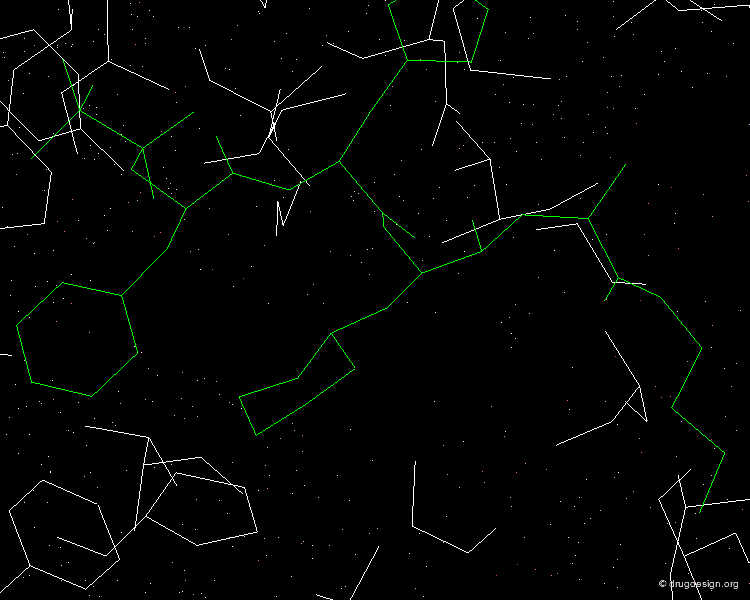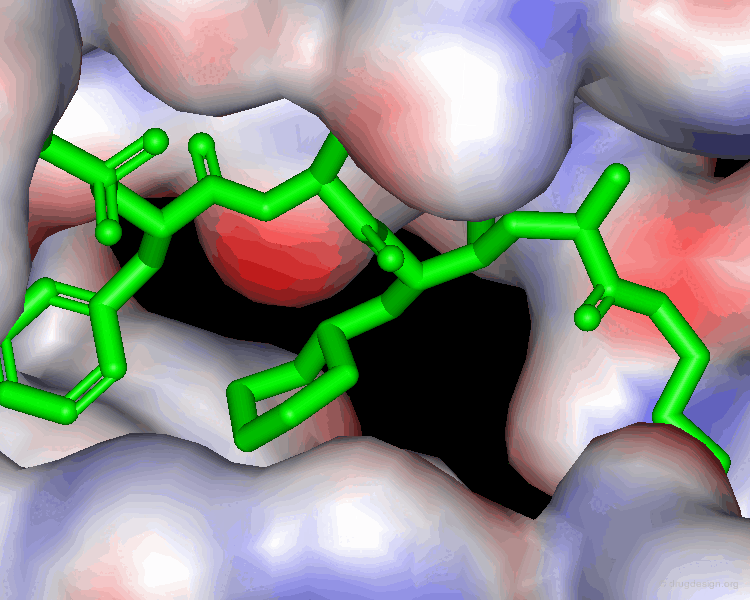
Predicting the Bioactive Conformation of CGP38560¶
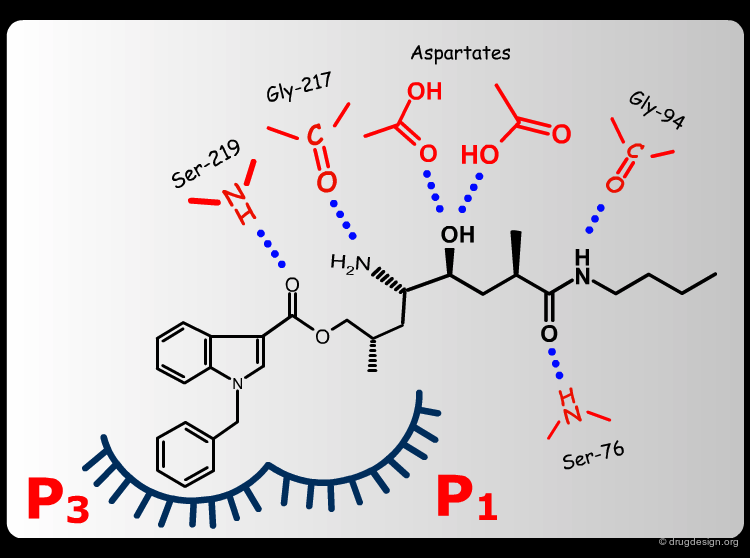
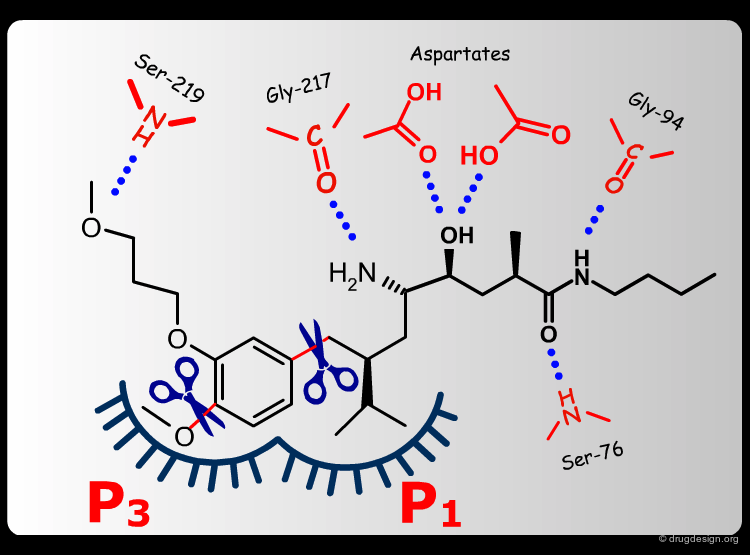
Extensive conformational and docking analyses were carried out to identify the Renin-CGP38560 complex with the lowest energy, which was assumed to correspond to the bioactive conformation of CGP38560. In this model the ligand binds into the sub-pockets P1, P2, P3 and P4 and forms six hydrogen bonds with the enzyme. It was estimated that P1 and P3 contributed to the binding more than P2 and P4.
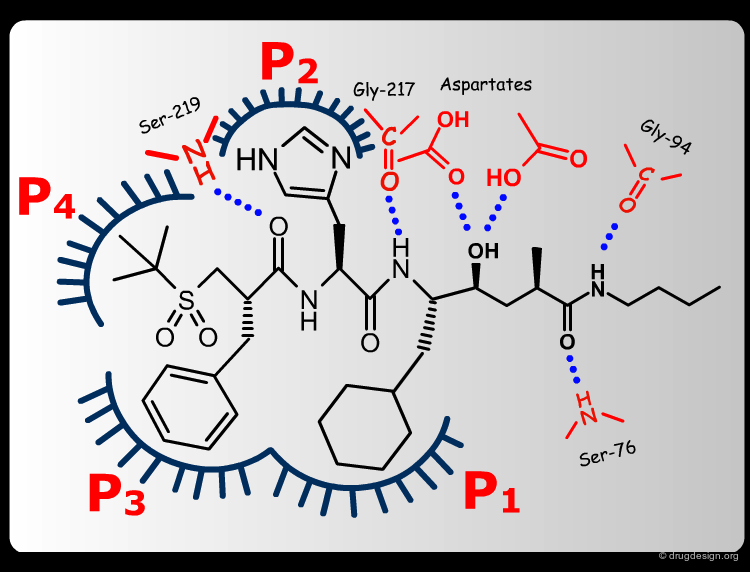
book
N.C. Cohen Trends in Medicinal Chemistry Elsevier Science Publisher 1989
The Design Strategy¶
The team came to the conclusion that in order to reduce the peptidic character of the CGP38560, part of its peptidic backbone had to be removed (as indicated in the figure below). By removing it, the Phe side-chain interacting at the P3 subsite would no longer be connected to the molecule but could be connected to the side-chain in P1. Analysis of binding requirements showed that although the P2 and P4 interactions could be sacrificed, a hydrogen-bond with the Serine-219 should be maintained.
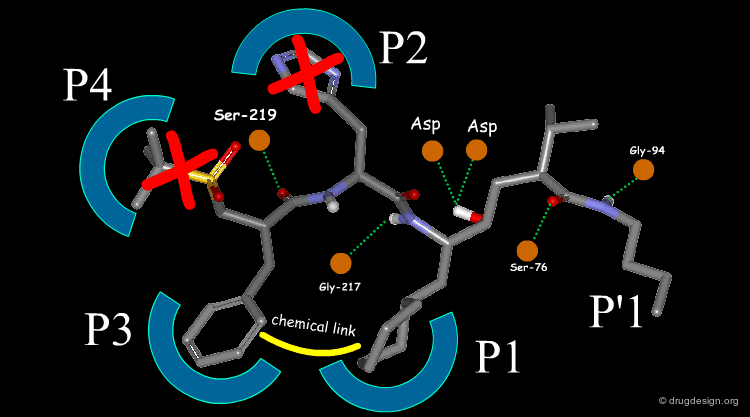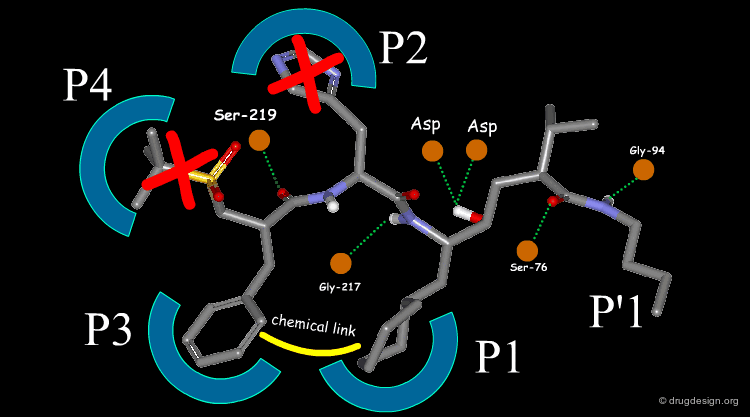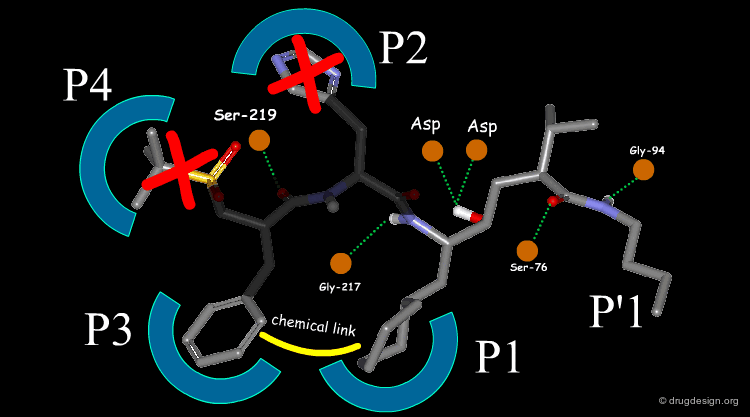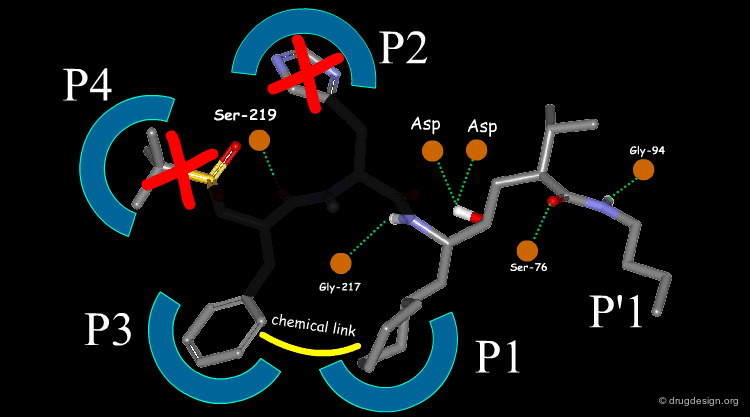
articles
Structure-based drug design and the discovery of aliskiren (Tekturna): perseverance and creativity to overcome a R and D pipeline challenge Nissim Claude Cohen Chem Biol Drug Des 70(6) 2007 10.1111/j.1747-0285.2007.00599.x
Finding a Feasible Scaffold¶
From the design strategy, the Novartis team defined their goal: to find a scaffold that would connect a group interacting at the P1 pocket, a group interacting at the P3 pocket and a group forming a hydrogen bond with Serine-219, to the rest of the molecule. The three teams of chemists started to search for suitable scaffolds based on their chemical feasibility and subsequently proposed hundreds of potential scaffolds to the modeler for consideration.

articles
Structure-based drug design and the discovery of aliskiren (Tekturna): perseverance and creativity to overcome a R and D pipeline challenge Nissim Claude Cohen Chem Biol Drug Des 70(6) 2007 10.1111/j.1747-0285.2007.00599.x
Criteria for Good Candidate Molecules¶
The modeler defined the criteria for a good candidate molecule: (1) a good overlap with the predicted bioactive conformation of the reference peptide, (2) the energy of the desired conformation of the candidate molecule had to be less than 1 kcal/mol above the global minimum of the molecule and (3) the existence of favorable interactions upon docking, including the formation of the six intermolecular hydrogen bonds.

articles
Structure-based drug design and the discovery of aliskiren (Tekturna): perseverance and creativity to overcome a R and D pipeline challenge Nissim Claude Cohen Chem Biol Drug Des 70(6) 2007 10.1111/j.1747-0285.2007.00599.x
The Parallel Design of Non-Peptide Renin Inhibitors¶
At the beginning most of the proposed scaffolds were unsuitable from a modeling point of view, but after some interactive educational work between the chemists and the modeler, the designs slowly came into focus. The three teams ended up with different scaffold designs: a tetrahydroquinoline, a phenoxy, an indole and a salicylamide series. The development of these four series' are described in more detail on the following pages.

The THQ Series¶
The first breakthrough was obtained by the Rasetti group with the tetrahydroquinoline (THQ) scaffold shown in the figure below; this molecule was active at the nanomolar range (and this was without optimization!). The superimposition (see button "Modeling and 3D design") shows a good overlap of the phenyl rings in P3. A gem-dimethyl group was introduced in P1 and a chemical spacer was designed to connect the two fragments. Note how the hydrogen bond with the Serine-219 was maintained.
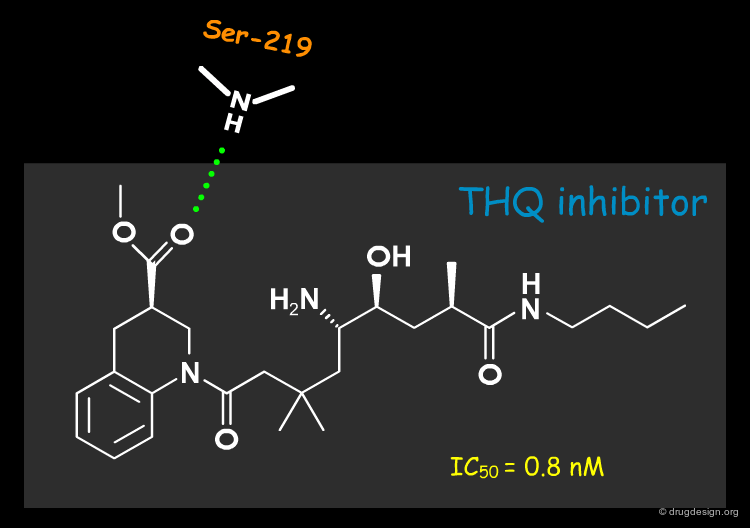


articles
Bioactive Hydroxyethylene Dipeptide Isosteres with Hydrophobic (P3-P1) Moieties, A Novel Strategy Towards Small Non-Peptide Renin Inhibitors V. Rasetti, N.C. Cohen, H. Rueger, R. Goeschke, J. Maibaum, F. Cumin, W. Fuhrer and J.M. Wood Bioorganic and Med. Chem. Letters 6 1996 10.1016/S0960-894X(96)00279-X
Validation of the Design Strategy¶
The good biological activity of the THQ molecule validated the project strategy. It is interesting to note that the THQ molecule was synthesized in three main steps and with each step the molecule gradually increased in potency. In the first step the linear anilino molecule (white) was synthesized (submicromolar activity), then the second ring (red) was introduced (IC50 of 10 -7M) and finally the ester group (yellow) was added in order to form a hydrogen-bond with the Serine residue.

articles
Structure-based drug design and the discovery of aliskiren (Tekturna): perseverance and creativity to overcome a R and D pipeline challenge Nissim Claude Cohen Chem Biol Drug Des 70(6) 2007 10.1111/j.1747-0285.2007.00599.x
The Phenoxy Series¶
The Goeschke research group worked in parallel on a second series with a phenyl-based scaffold. Analyses of this series showed that the phenyl ring did not align well with the Phe residue of the CGP38560; therefore a tert-butyl group was added at the para-position to extend the molecule and occupy the Phe position in the P3 pocket. The isopropyl moiety at P1 mimicked the cyclohexane group of the peptide well. This initial scaffold had an IC50 of 0.1 µM.



articles
Novel 2,7-Dialkyl Substituted 5(S)-Amino-4(S)-Hydroxy-8-Phenyl-Octanecarboxamide Transition State Peptidomimetics are Potent and Orally Active Inhibitors of Human Renin Richard Goeschke, Vittorio Rasetti, Nissim-Claude Cohen, Joseph Rahuel, Hans-Peter Baum, Peter Forgiarini, Christian R. Schnell, Markus G. Gruetter, Stefan Stutz, Grety Rihs, Trixie Wagner, Walter Fuhrer, Frederic Cumin, Jeanette M. Wood, Juergen Maibaum. J Med Chem 50 (20) 2007 10.1021/jm070314y
Structural Modification of the P2' Position of 2,7-Dialkyl Substituted 5(S)-Amino-4(S)-Hydroxy-8-Phenyl-Octanecarboxamides: The Discovery of Aliskiren, A Potent Non-Peptide Human Renin Inhibitor Active After Once Daily Dosing in Marmosets Juergen Maibaum, Stefan Stutz, Richard Goeschke, Pascal Rigollier, Yasuchika Yamaguchi, Frederic Cumin, Joseph Rahuel, Hans-Peter Baum, Nissim-Claude Cohen, Christian R. Schnell, Walter Fuhrer, Markus G. Gruetter, Walter Schilling, Jeanette M. Wood J Med Chem 50 (20) 2007 10.1021/jm070316i
Structure-based drug design and the discovery of aliskiren (Tekturna): perseverance and creativity to overcome a R and D pipeline challenge Nissim Claude Cohen Chem Biol Drug Des 70(6) 2007 10.1111/j.1747-0285.2007.00599.x
Novel 2,7 Dialkyl Substituted 5(S)-Amino-4(S)-Hydroxy-8-Phenyl-Octanecarboxamide Transition State Peptidomimetics are Potent and Orally Active Inhibitors of Human Renin R. Goeschke, V. Rasetti, N.C. Cohen, J. Rahuel, M. Gruetter, S. Stutz, H. Rueger, J.M. Wood, J. Maibaum XVth Meeting of the European Federation of Medicinal Chemistry, Edinburg
Sept. 6- 10, 1998
Design and Syntheses of Novel 2,7Dialkyl substituted 5(S)Amino 4(S)Hydroxy-8Phenyl- Octanecarboxamides-as in vitro Potent Peptidomimetic Inhibitors of Humain Renin R. Goeschke, N.C. Cohen, J.M. Wood and J. Maibaum Bioorganic and Med. Chem. Letters 7 1997
book
J. Maibaum, V. Rasetti, H. Rueger, N.C. Cohen, R. Goechke, R. Mah, J. Rahuel, M. Gruetter, F. Cumin and J.M. Wood Medicinal Chemistry Today and Tomorrow Blackwell Science 1997
Optimization of the Phenoxy Lead¶
The tert-butyl was replaced by a methoxy because it was synthetically more accessible and retained good biological activity. Modeling analyses showed that in order to create the hydrogen-bond with Serine-219, a substitution should be made at position 3' of the phenyl ring, which was additionally confirmed by superimposing the molecule with the THQ lead (Sup). The substituted ester "A" and the dialkoxy "B" (see "Nanomolar Leads") led to molecules with nanomolar activities. Aliskiren is a direct descendant of the structures of the phenoxy series.

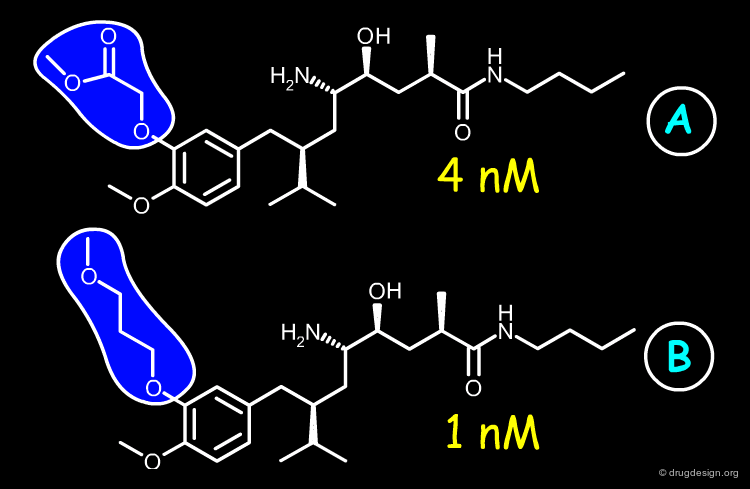

articles
Structure-based drug design and the discovery of aliskiren (Tekturna): perseverance and creativity to overcome a R and D pipeline challenge Nissim Claude Cohen Chem Biol Drug Des 70(6) 2007 10.1111/j.1747-0285.2007.00599.x
The Indole Series¶
The Maibaum research group worked in parallel on a third series with an indole-based scaffold. A benzyl group was added to the indole heterocycle to mimic the Phe residue of the peptide located in P3. The carbonyl group of the ester was expected to establish a hydrogen-bond with Ser-219. The molecule shown here was synthesized and had an IC50 of 3nM.


The Salicylamide Series¶
After the discovery of the three series, their X-ray complexes with renin were determined and confirmed the poor overlap of the phenyl ring of the phenoxy structure with the Phe residue of CGP38560 that was predicted by modeling. The modeler raised the two following questions: (1) what is the preferred location for a phenyl ring in the P3 pocket? and (2) how could the phenoxy scaffold be modified to position its phenyl ring at the preferred location?

articles
Design and Synthesis of Novel, Fully Non-Peptide Transition State Mimic Renin Inhibitors bearing an O-alkyl Substitued Salicylamide (P3-sp-P3)-Moiety with High Oral in vivo Potency J. Maibaum, N.C. Cohen, J. Rahuel, Ch. Schnell, H-P. Baum, P. Rigollier, W. Schilling, J. Wood XVth Meeting of the European Federation of Medicinal Chemistry
Edinburg, Sept. 6-10, 1998
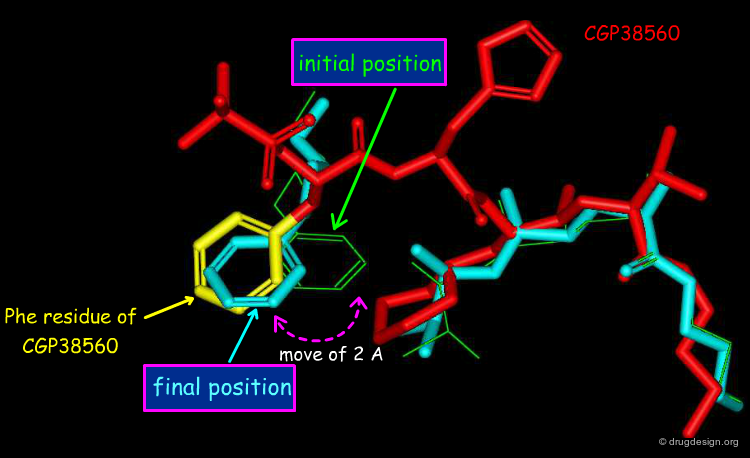
A Docking Experiment¶
To answer the first question, the dialkoxy-phenyl moiety of the phenoxy inhibitor was disconnected from the second part of the molecule, and the two disconnected parts were then docked simultaneously - each part being free to bind optimally to the recently determined X-ray structure of the enzyme. The docking simulation revealed that the aromatic ring of the phenyl-dialkoxy moiety moved approximately 2Å away from the other moiety, and reached the exact position occupied by the phenyl ring of the Phe residue of CGP38560.
![]()
Design of the Salicylamide Molecule¶
Sitting in front of the screen, with the model of the disconnected docked moieties into the 3D structure of renin, Cohen and Maibaum searched for spacers that would assemble the two parts in one single molecule. The carboxamidomethylene (CO-NH-CH2) spacer was selected as the best solution; the first molecule that was prepared proved to be active at the nanomolar level. This led to a fourth family of inhibitors: the salicylamide series.



articles
Design and Synthesis of Novel, Fully Non-Peptide Transition State Mimic Renin Inhibitors bearing an O-alkyl Substitued Salicylamide (P3-sp-P3)-Moiety with High Oral in vivo Potency J. Maibaum, N.C. Cohen, J. Rahuel, Ch. Schnell, H-P. Baum, P. Rigollier, W. Schilling, J. Wood XVth Meeting of the European Federation of Medicinal Chemistry
Edinburg, Sept. 6-10, 1998
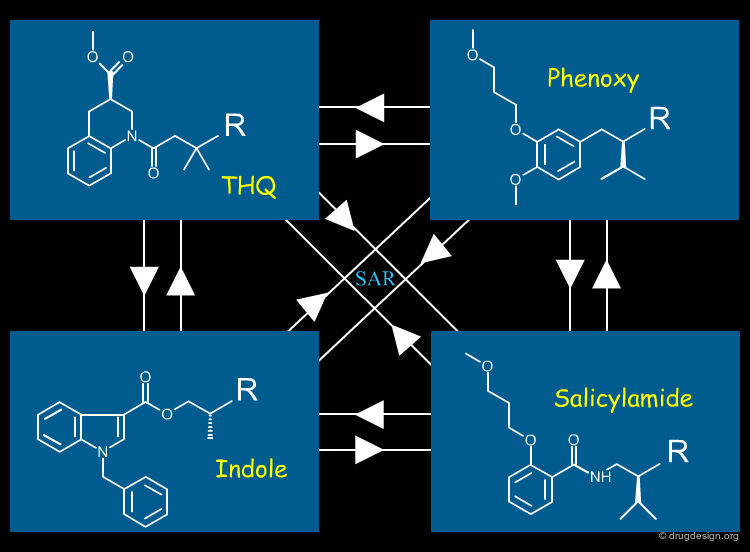
Transferrable SAR's¶
Although the four scaffolds were structurally unrelated, the 3D modeling strategy allowed any structure-activity relationship result found in one series to be successfully applied to the other series' and this was used extensively throughout the design process.
Example of Transferrable SAR's¶
To illustrate the successful transfer of SAR's between series, one example is given here. In order to form a hydrogen bond with Ser-219, the dialkoxy tail of the phenoxy series had to be placed at the meta position of the benzene ring. When transferring this knowledge to the salicylamide series, a 2D representation of the molecules was insufficient to deduce the correct positioning of the dialkoxy tail. Only by viewing the molecules in 3D can we see that the dialkoxy chain has to be copied to the ortho position of the salicylamide benzene ring to ensure good 3D mimicry.
![]()

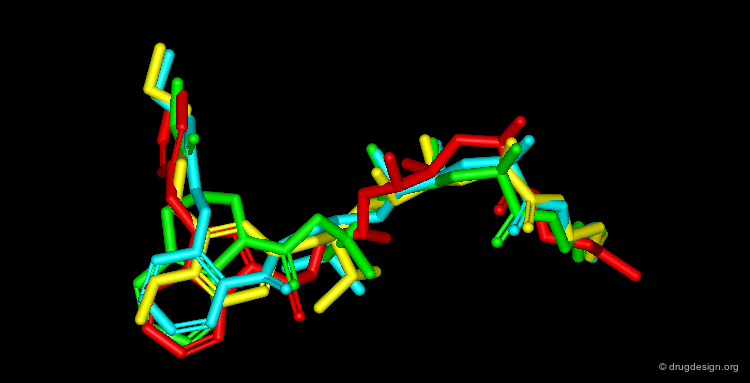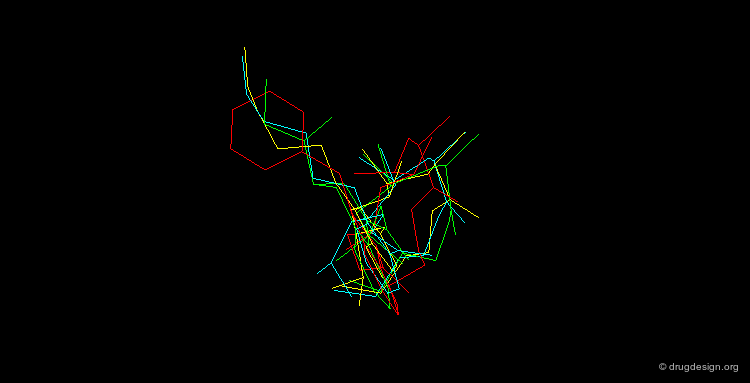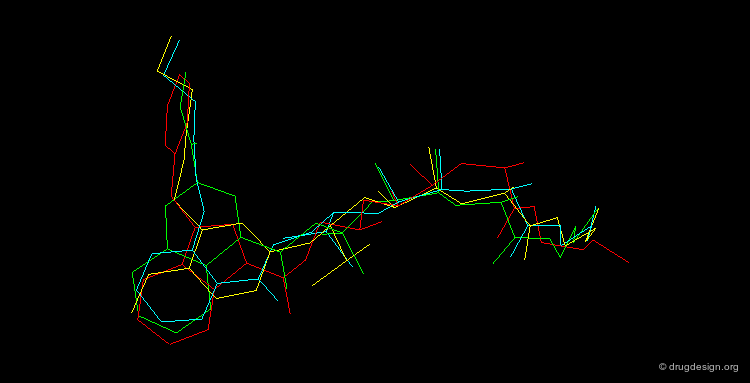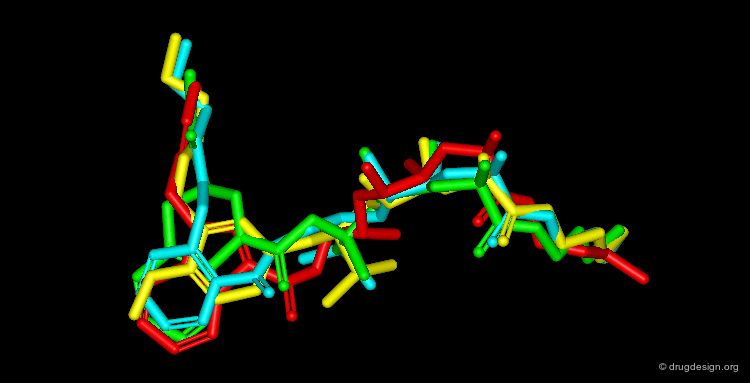
Four Unrelated Lead Compounds¶
By November 1992 after 3 years of extensive work the team had discovered four structurally unrelated lead compounds each active at low nanomolar range and each had the potential to be developed into a therapeutic renin inhibitor: a THQ compound, a phenoxy compound, an indole compound and a salicylamide compound. A superimposition of the four compounds is illustrated below; it summarizes in a single view the successful achievements obtained after several years of intensive effort.
articles
Novel 2,7-Dialkyl Substituted 5(S)-Amino-4(S)-Hydroxy-8-Phenyl-Octanecarboxamide Transition State Peptidomimetics are Potent and Orally Active Inhibitors of Human Renin Richard Goeschke, Vittorio Rasetti, Nissim-Claude Cohen, Joseph Rahuel, Hans-Peter Baum, Peter Forgiarini, Christian R. Schnell, Markus G. Gruetter, Stefan Stutz, Grety Rihs, Trixie Wagner, Walter Fuhrer, Frederic Cumin, Jeanette M. Wood, Juergen Maibaum. J Med Chem 50 (20) 2007 10.1021/jm070314y
Structural Modification of the P2' Position of 2,7-Dialkyl Substituted 5(S)-Amino-4(S)-Hydroxy-8-Phenyl-Octanecarboxamides: The Discovery of Aliskiren, A Potent Non-Peptide Human Renin Inhibitor Active After Once Daily Dosing in Marmosets Juergen Maibaum, Stefan Stutz, Richard Goeschke, Pascal Rigollier, Yasuchika Yamaguchi, Frederic Cumin, Joseph Rahuel, Hans-Peter Baum, Nissim-Claude Cohen, Christian R. Schnell, Walter Fuhrer, Markus G. Gruetter, Walter Schilling, Jeanette M. Wood J Med Chem 50 (20) 2007 10.1021/jm070316i
Structure-based drug design and the discovery of aliskiren (Tekturna): perseverance and creativity to overcome a R and D pipeline challenge Nissim Claude Cohen Chem Biol Drug Des 70(6) 2007 10.1111/j.1747-0285.2007.00599.x
Design and Syntheses of Novel 2,7 Dialkyl substituted 5(S)Amino 4(S)Hydroxy-8 Phenyl- Octanecarboxamides-as in vitro Potent Peptidomimetic Inhibitors of Humain Renin R. Goeschke, N.C. Cohen, J.M. Wood and J. Maibaum Bioorganic and Med. Chem. Letters 7 1997
Bioactive Hydroxyethylene Dipeptide Isosteres with Hydrophobic (P3-P1) Moieties, A Novel Strategy Towards Small Non-Peptide Renin Inhibitors V. Rasetti, N.C. Cohen, H. Rueger, R. Goeschke, J. Maibaum, F. Cumin, W. Fuhrer and J.M. Wood Bioorganic and Med. Chem. Letters 6 1996 10.1016/S0960-894X(96)00279-X
Structure-based design of aliskiren, a novel orally effective renin inhibitor Jeanette M. Wood, Juergen Maibaum, Joseph Rahuel, Marcus G. Gruetter, Nissim-Claude Cohen, Vittorio Rasetti, Heinrich Rueger, Richard Goeschke, Stefan Stutz, Walter Fuhrer, Walter Schilling, Pascal Rigollier, Yasuchika Yamaguchi, Frederic Cumin, Hans-Peter Baum, Christian R. Schnell, Peter Herold, Robert Mah, Chris Jensen, Eoin O'Brien, Alice Stanton and Martin P. Bedigian Biochemical and Biophysical Research Communications 308 2003 10.1016/S0006-291X(03)01451-7
book
J. Maibaum, V. Rasetti, H. Rueger, N.C. Cohen, R. Goechke, R. Mah, J. Rahuel, M. Gruetter, F. Cumin and J.M. Wood Medicinal Chemistry Today and Tomorrow Blackwell Science 1997
Browser of the Novartis Renin Inhibitor Leads¶

articles
Novel 2,7-Dialkyl Substituted 5(S)-Amino-4(S)-Hydroxy-8-Phenyl-Octanecarboxamide Transition State Peptidomimetics are Potent and Orally Active Inhibitors of Human Renin Richard Goeschke, Vittorio Rasetti, Nissim-Claude Cohen, Joseph Rahuel, Hans-Peter Baum, Peter Forgiarini, Christian R. Schnell, Markus G. Gruetter, Stefan Stutz, Grety Rihs, Trixie Wagner, Walter Fuhrer, Frederic Cumin, Jeanette M. Wood, Juergen Maibaum. J Med Chem 50 (20) 2007
Structural Modification of the P2' Position of 2,7-Dialkyl Substituted 5(S)-Amino-4(S)-Hydroxy-8-Phenyl-Octanecarboxamides: The Discovery of Aliskiren, A Potent Non-Peptide Human Renin Inhibitor Active After Once Daily Dosing in Marmosets Juergen Maibaum, Stefan Stutz, Richard Goeschke, Pascal Rigollier, Yasuchika Yamaguchi, Frederic Cumin, Joseph Rahuel, Hans-Peter Baum, Nissim-Claude Cohen, Christian R. Schnell, Walter Fuhrer, Markus G. Gruetter, Walter Schilling, Jeanette M. Wood J Med Chem 50 (20) 2007
Structure-based drug design and the discovery of aliskiren (Tekturna): perseverance and creativity to overcome a R and D pipeline challenge Nissim Claude Cohen Chem Biol Drug Des 70(6) 2007
Design and Syntheses of Novel 2,7 Dialkyl substituted 5(S)Amino 4(S)Hydroxy-8 Phenyl- Octanecarboxamides-as in vitro Potent Peptidomimetic Inhibitors of Humain Renin R. Goeschke, N.C. Cohen, J.M. Wood and J. Maibaum Bioorganic and Med. Chem. Letters 7 1997
Bioactive Hydroxyethylene Dipeptide Isosteres with Hydrophobic (P3-P1) Moieties, A Novel Strategy Towards Small Non-Peptide Renin Inhibitors V. Rasetti, N.C. Cohen, H. Rueger, R. Goeschke, J. Maibaum, F. Cumin, W. Fuhrer and J.M. Wood Bioorganic and Med. Chem. Letters 6 1996
Structure-based design of aliskiren, a novel orally effective renin inhibitor Jeanette M. Wood, Juergen Maibaum, Joseph Rahuel, Marcus G. Gruetter, Nissim-Claude Cohen, Vittorio Rasetti, Heinrich Rueger, Richard Goeschke, Stefan Stutz, Walter Fuhrer, Walter Schilling, Pascal Rigollier, Yasuchika Yamaguchi, Frederic Cumin, Hans-Peter Baum, Christian R. Schnell, Peter Herold, Robert Mah, Chris Jensen, Eoin O'Brien, Alice Stanton and Martin P. Bedigian Biochemical and Biophysical Research Communications 308 2003
book
J. Maibaum, V. Rasetti, H. Rueger, N.C. Cohen, R. Goechke, R. Mah, J. Rahuel, M. Gruetter, F. Cumin and J.M. Wood Medicinal Chemistry Today and Tomorrow Blackwell Science 1997
From Initial Lead to Aliskiren¶
An optimization program was carried out which involved the synthesis of more that 1400 analogs of all four series. One of the phenoxy analogs was selected for further development (later named Aliskiren) as it possessed the best physicochemical and pharmacokinetic properties. The compound entered into clinical trials in 1999 and was approved by the FDA in March 2007. Aliskiren trade names include Tekturna (USA) and Rasilez (Europe).


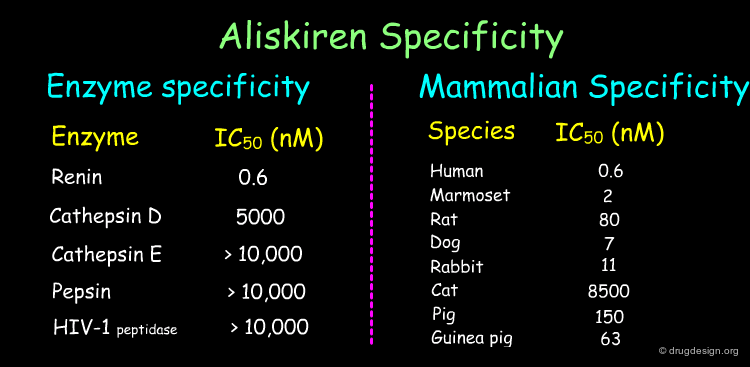
articles
Novel 2,7-Dialkyl Substituted 5(S)-Amino-4(S)-Hydroxy-8-Phenyl-Octanecarboxamide Transition State Peptidomimetics are Potent and Orally Active Inhibitors of Human Renin Richard Goeschke, Vittorio Rasetti, Nissim-Claude Cohen, Joseph Rahuel, Hans-Peter Baum, Peter Forgiarini, Christian R. Schnell, Markus G. Gruetter, Stefan Stutz, Grety Rihs, Trixie Wagner, Walter Fuhrer, Frederic Cumin, Jeanette M. Wood, Juergen Maibaum. J Med Chem 50 (20) 2007
Structural Modification of the P2' Position of 2,7-Dialkyl Substituted 5(S)-Amino-4(S)-Hydroxy-8-Phenyl-Octanecarboxamides: The Discovery of Aliskiren, A Potent Non-Peptide Human Renin Inhibitor Active After Once Daily Dosing in Marmosets Juergen Maibaum, Stefan Stutz, Richard Goeschke, Pascal Rigollier, Yasuchika Yamaguchi, Frederic Cumin, Joseph Rahuel, Hans-Peter Baum, Nissim-Claude Cohen, Christian R. Schnell, Walter Fuhrer, Markus G. Gruetter, Walter Schilling, Jeanette M. Wood J Med Chem 50 (20) 2007
Structure-based design of aliskiren, a novel orally effective renin inhibitor Jeanette M. Wood, Juergen Maibaum, Joseph Rahuel, Marcus G. Gruetter, Nissim-Claude Cohen, Vittorio Rasetti, Heinrich Rueger, Richard Goeschke, Stefan Stutz, Walter Fuhrer, Walter Schilling, Pascal Rigollier, Yasuchika Yamaguchi, Frederic Cumin, Hans-Peter Baum, Christian R. Schnell, Peter Herold, Robert Mah, Chris Jensen, Eoin O'Brien, Alice Stanton and Martin P. Bedigian Biochemical and Biophysical Research Communications 308 2003
The Aliskiren Project Timeline¶
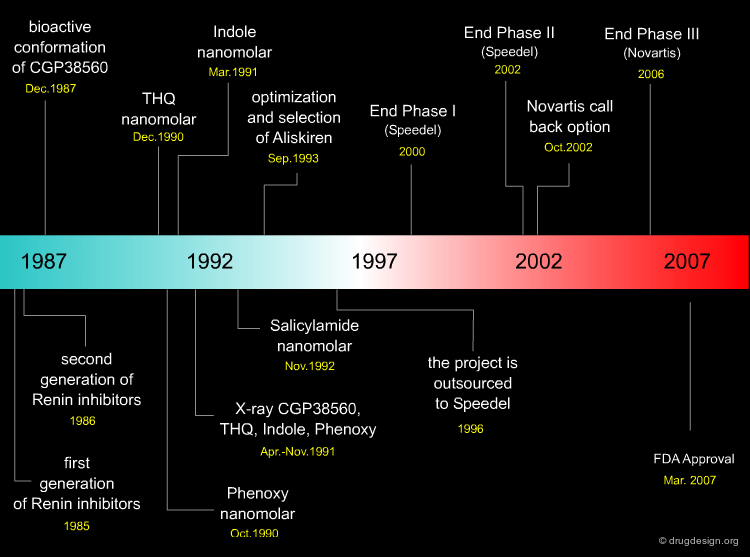
What Made the Success of the Project Possible?¶
Many pharmaceutical companies have been involved in the discovery of Renin inhibitors for more than 20 years, investing millions of hours of scientific effort, intensive teamwork, human creativity and money into projects that have failed to produce suitable drugs. What enabled Novartis to successfully meet this challenge? This question is addressed in the following three pages.

articles
Structure-based drug design and the discovery of aliskiren (Tekturna): perseverance and creativity to overcome a R and D pipeline challenge Nissim Claude Cohen Chem Biol Drug Des 70(6) 2007 10.1111/j.1747-0285.2007.00599.x
The Incorporation of Modeling¶
Novartis luckily was not put off by the failure of CGP38560 in clinical trials and had the foresight to incorporate the expertise of a molecular modeler who gave the research group the ability to start tackling the problem from a modeling perspective.
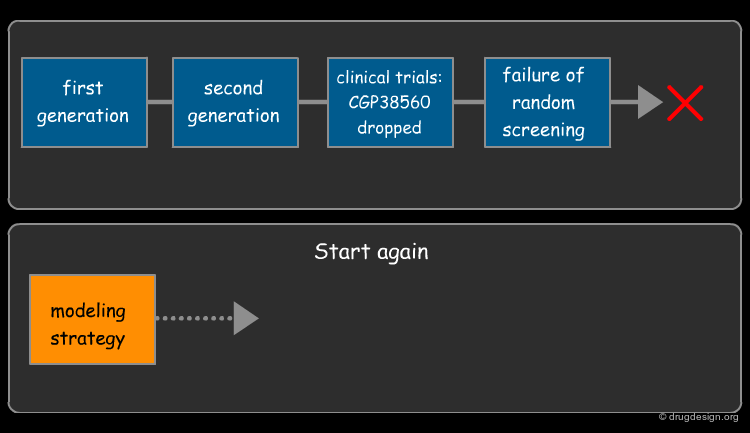
Modeling - The Key to Aliskiren's Success¶
Modeling was the key to Aliskiren's success; based only on the homology model of renin, the team predicted the CGP38560 bioactive conformation, analyzed the binding requirements, defined a strategy, designed molecules and rapidly identified lead compounds with nanomolar activity without the need for much optimization.

articles
Structure-based drug design and the discovery of aliskiren (Tekturna): perseverance and creativity to overcome a R and D pipeline challenge Nissim Claude Cohen Chem Biol Drug Des 70(6) 2007 10.1111/j.1747-0285.2007.00599.x
Historical Document¶
This achievement impressed the Novartis management and they asked the team to write a signed and dated document summarizing their structure-based predictions to serve as a basis for comparison with experimental X-ray data when these became available. These predictions were confirmed experimentally one year later.
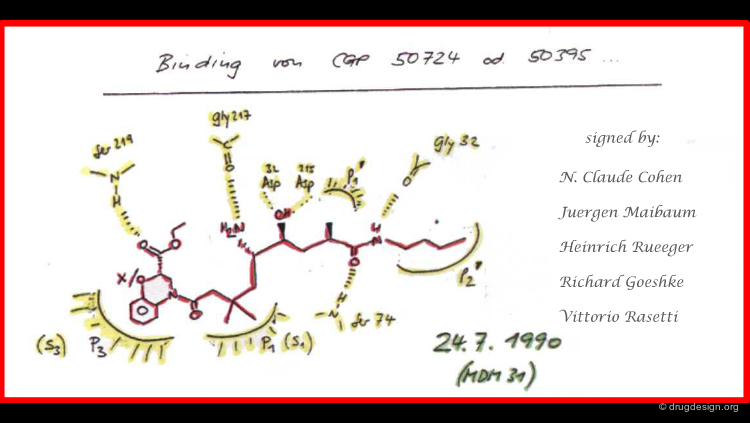
articles
Structure-based drug design and the discovery of aliskiren (Tekturna): perseverance and creativity to overcome a R and D pipeline challenge Nissim Claude Cohen Chem Biol Drug Des 70(6) 2007 10.1111/j.1747-0285.2007.00599.x
Good Teamwork¶
Another key factor that led to the success of this project was the exemplary co-operation between the medicinal chemists and the modeler. Daily collaborative meetings (the MDM - "Molecular Design Meetings") were held and focused on design issues. The meetings were constructive as they strengthened the team as a unit and gave each member a solid grasp of both the individual and group objectives thus allowing the team to converge towards many successful designs.

articles
Structure-based drug design and the discovery of aliskiren (Tekturna): perseverance and creativity to overcome a R and D pipeline challenge Nissim Claude Cohen Chem Biol Drug Des 70(6) 2007 10.1111/j.1747-0285.2007.00599.x
Following the Discovery¶

X-rays of Complex with CGP38560¶
In 1991 X-ray studies were carried out on the reference CGP38560 peptide. The modeling predictions of the bioactive conformation made in 1987 proved to be correct and gave the research team a four-year head start on the project. The difference in the position of the Histidine moiety is of minor importance and did not affect the project.


articles
Structure Based Drug Design: the Discovery of Novel Non-Peptide Orally Active Inhibitors of Human Renin J. Rahuel, V. Rasetti, J. Maibaum, H. Rueger, R. Goeschke, N.C. Cohen, S. Stutz, F. Cumin, W. Fuhrer, J.M. Wood and M. Gruetter Chemistry and Biology 7 2000
Renin inhibition by substituted piperidines: a novel paradigm for the inhibition of monomeric aspartic proteinases? Oefner C, Binggeli A, Breu V, Bur D, Clozel JP, D'Arcy A, Dorn A, Fischli W, Gruninger F, Guller R, Hirth G, Marki H, Mathews S, M ller M, Ridley RG, Stadler H, Vieira E, Wilhelm M, Winkler F and Wostl W Chem. Biol 6 1999
book
N. Claude Cohen Trends in Medicinal Chemistry Elsevier Science Publisher 1989
X-ray Determination of Lead Inhibitors¶
X-ray determinations were carried out with all the lead compounds discovered. They confirmed the accuracy of the predictions made on the basis of modeling studies, with however one surprise concerning the indole inhibitor, as detailed in the following page.
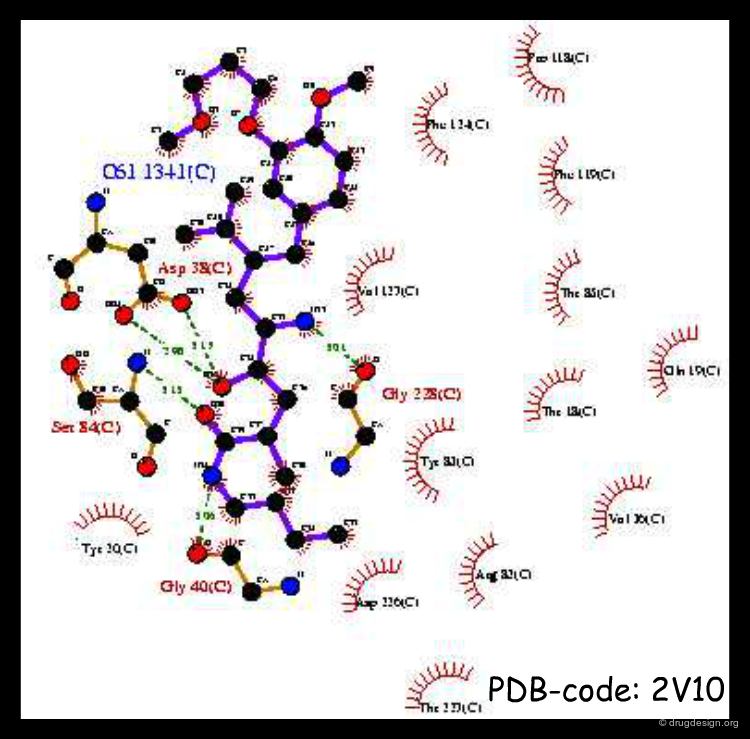
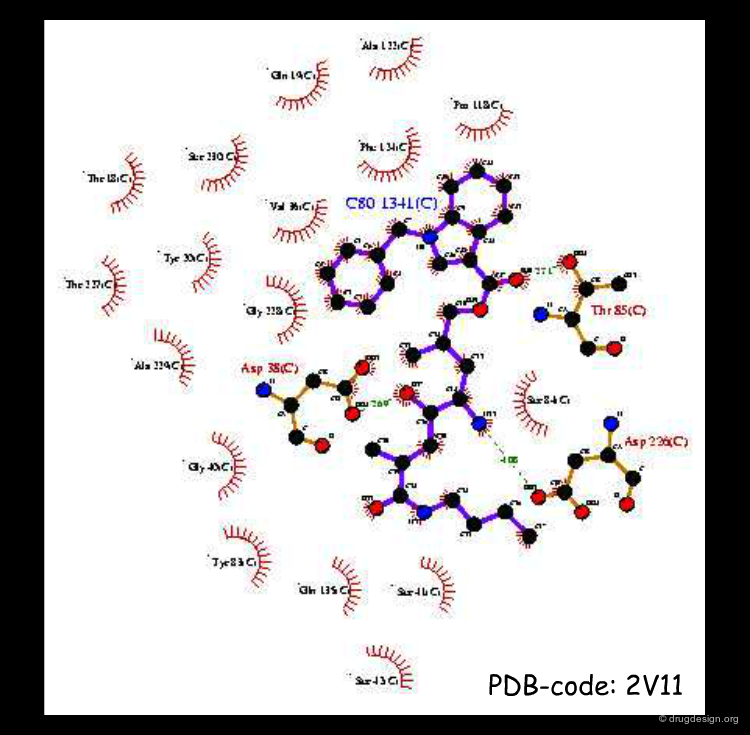


articles
Structure Based Drug Design: the Discovery of Novel Non-Peptide Orally Active Inhibitors of Human Renin J. Rahuel, V. Rasetti, J. Maibaum, H. Rueger, R. Goeschke, N.C. Cohen, S. Stutz, F. Cumin, W. Fuhrer, J.M. Wood and M. Gruetter Chemistry and Biology 7 2000
The Indole X-ray¶
The X-ray of the indole inhibitor revealed that its binding mode was not as predicted and the indole heterocyle was in fact inverted, with the benzyl group pointing towards the S3sp sub-pocket. The reason for the indole's good potency was that its benzyl moiety actually interacted with the S3sp sub-pocket. This was the first hint that the hydrogen bond formed with the Serine-219 was not mandatory.

articles
Structure Based Drug Design: the Discovery of Novel Non-Peptide Orally Active Inhibitors of Human Renin J. Rahuel, V. Rasetti, J. Maibaum, H. Rueger, R. Goeschke, N.C. Cohen, S. Stutz, F. Cumin, W. Fuhrer, J.M. Wood and M. Gruetter Chemistry and Biology 7 2000
The S3sp sub-Pocket¶
The X-ray structures of the complexes revealed the existence of a sub-pocket that is not exploited by the natural substrate or the peptide-like inhibitors. The additional side chains (e.g. dialkoxy tail) designed to interact with Ser-219 luckily entered this sub-pocket. The strategic decision to keep the hydrogen-bond with Ser-219 proved to be a key element in the success of the project.

articles
Structure Based Drug Design: the Discovery of Novel Non-Peptide Orally Active Inhibitors of Human Renin J. Rahuel, V. Rasetti, J. Maibaum, H. Rueger, R. Goeschke, N.C. Cohen, S. Stutz, F. Cumin, W. Fuhrer, J.M. Wood and M. Gruetter Chemistry and Biology 7 2000
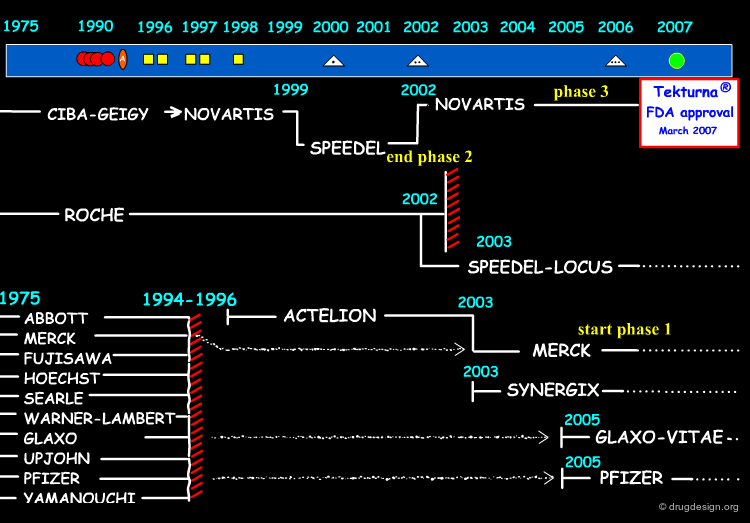
Other Work on Drugs in this Class¶
The diagram here gives us a global perspective of the effort deployed by the various pharmaceutical companies over the years in the search for renin inhibitors. The Novartis story is represented on the top. Note that the Phase 1 and 2 clinical trials were carried out by Speedel and Novartis exercised their call-back option in 2002. Companies like Merck, Pfizer and Glaxo have recently shown renewed interest and reactivated their renin inhibitor projects.
Copyright © 2024 drugdesign.org