Principles of Structure-Based Design¶
Info
The different possibilities for designing molecules in a structure-based design approach are presented and discussed. The different steps involved in the design process are presented and illustrated on a success story.
Number of Pages: 92 (±2 hours read)
Last Modified: January 2004
Prerequisites: Principles of Rational Drug Design, Structure-Based Drug Design: Analysis
Introduction¶
Design of Drug Candidates: An Iterative Process¶
The design of new ligands is carried out as a step-by-step procedure. The state-of-the art design process is based in large part, on a good understanding of the molecular recognition of protein-ligand complexes relying upon analogies to other systems and using advanced computerized molecular design programs.

Steps in Structure-Based Drug Design¶
The steps used in structure-based drug design for designing new lead compounds are illustrated in the following diagram.

Small Changes Can Produce Huge Effects¶
When structural changes are made that modify the energy of interaction of a ligand with its receptor, one can observe a large change in the biological properties. For example the SB203580 molecule has a Ki of 100 nM on the protein kinase p38. However the molecule looses its activity if residue Thr-106 is mutated by Met-106. The same molecule is entirely inactive on the kinase ERK2, however if the residue Glu-105 of the protein is mutated by Thr-105, the Ki of the molecule becomes 13 nM. The following pages will provide a 3D interpretation of these observations.

articles
Three-Dimensional Structures of Drug-resistant Mutants of Human Rhinivirus 14 Badger J, Krishnaswamy S, Kremer MJ, Oliveira MA, Rossmann, Heinz BA, Rueckert RR, Dutko FJ and McKinlay MA J. Mol. Biol. 207 1989
A Single Amino Acid Substitution Makes ERK2 Susceptible to Pyridinyl Imidazole Inhibitors of p38 MAP kinase Fox T, Coll JT, Xie X, Ford PJ, Germann UA, Porter MD, Pazhanisamy S, Fleming MA, Galullo V, Su MS, Wilson KP Protein Sci. 7 1998
Structural Basis of Inhibitor Selectivity in MAP Kinases Wang Z, Canagarajah BJ, Boehm JC, Kassisa S, Cobb MH, Young PR, Abdel-Meguid S, Adams JL, Goldsmith EJ Structure 6 1998
p38 Wild¶
The crystallographic structure of the complex of the inhibitor SB203580 with the p38 kinase reveals good complementary features between the inhibitor and the protein.

p38 Mutant¶
Replacing in the X-ray structure residue Thr-106 by a Met-106 reveals the existence of strong steric repulsions between the mutated protein and SB203580. This explains why SB203580 is not anymore active on the mutated protein.
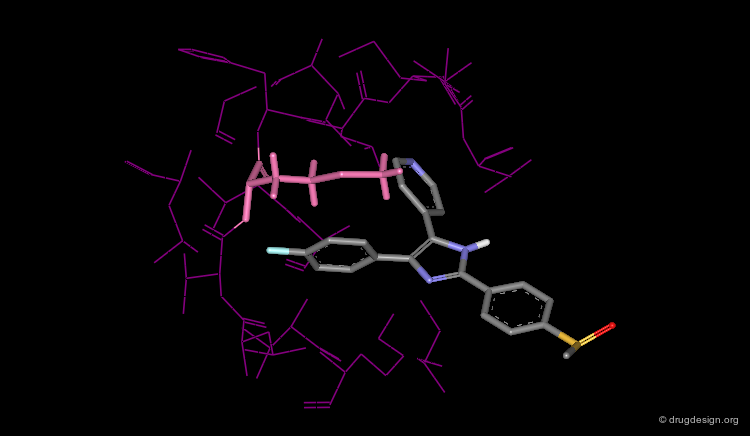
ERK-2 Wild¶
There is no complex between SB203580 with the wild ERK2 kinase. The following model was constructed using the crystallographic structure of the complex with the ERK2-(Q105T) mutant. It shows strong repulsions between SB203580 and the Glu-105 residue and explains why the compound is inactive on ERK2.

ERK-2 Mutant¶
The crystallographic structure of the complex of SB203580 with the mutated ERK2 (Q105T) kinase reveals favorable interactions between the inhibitor and the protein. This example shows how a single point mutation can transform an inactive molecule into a nanomolar inhibitor. Small structural modifications can greatly affect the potency of an inhibitor, this can be well exploited for achieving potency and specificity.

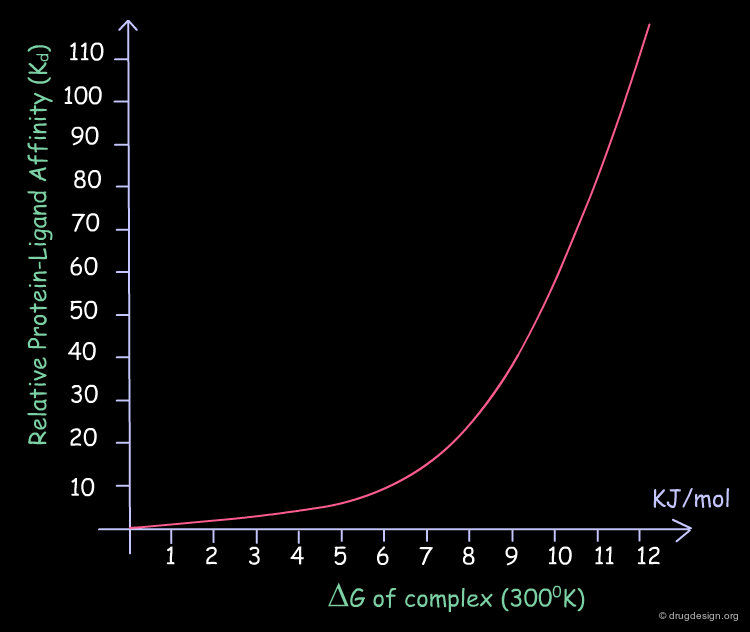
Increasing Biological Activity¶
Each small amount that is gained in the binding energy of a ligand is translated into significant improvements in its biological activity. The following table illustrates the relative values of Kd as a function of the binding energies, at 300°K.
Beginning the Design Phase¶
Once the phase of analysis is complete, the design phase can start. One has to identify candidate scaffolds with appropriate substituents that can ensure enhanced interactions with selected sites of the protein. In the case of the optimization of a known series, the information is used to design new analogs.

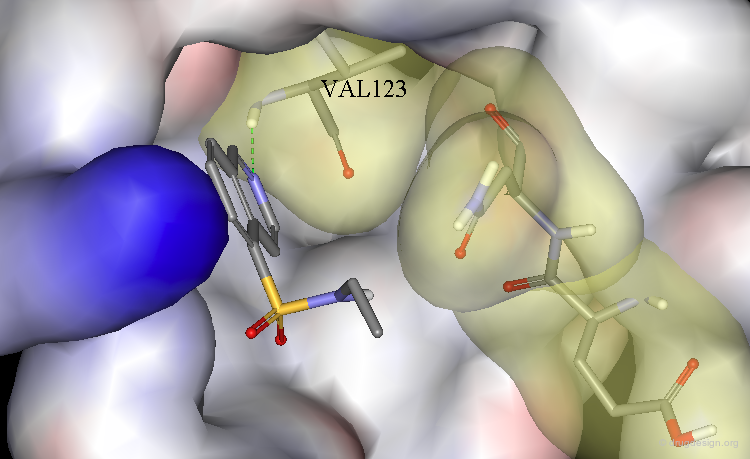
A Simple Example of Design¶
A molecule active at the micromolar range has been discovered and the X-ray structure of the complex of this molecule with the target protein has been solved. Following is an example of analog designed from the X-ray data.
articles
Crystal structures of catalytic subunit of cAMP-dependent protein kinase in complex with isoquinolinesulfonyl protein kinase inhibitors H7, H8, and H89 Structural implications for selectivity Engh RA, Girod A, Kinzel V, Huber R, Bossemeyer D J. Biol. Chem. 271 1996
Extension of the Molecule to Form another H-Bond¶
The X-ray structure reveals the binding mode of the micromolar molecule. On the basis of this information one could expect to obtain a more potent compound by making the amino-analog of the molecule. As can be seen on the view, the amino moiety creates hydrogen bonds with two backbone carbonyls of the target protein.

Checking the Validity of the Design¶
The next step is to test the biological activity of the molecule to see if the design concept was structurally correct. This information could provide answers to such questions as: If the molecule was potent, was it potent for the correct reasons built into the design? If the compound was weaker than expected, how and why did the concept design fail?

Definition of Docking¶
Given the 3D structure of a protein target, compounds can be designed to fit in a cavity, which is called "docking". Starting with an approximate orientation of a ligand into the active site, the docking process modifies its position and conformation in order to have a maximum of favorable intermolecular interactions. The treatment ends when a minimum of energy is obtained for the complex.

articles
A Good Ligands is Hard to Find: Automated Docking Methods Blaney JM and Dixon JS Perspect. Drug Discov. Design 1 1993
Accommodating Protein Flexibility in Computational Drug Design Carlson HA and McCammon JA Mol Pharmacol. 57 2000
Docking Flexible Ligands to Macromolecular Receptors by Molecular Shape DesJarlais RL, Sheridan RP, Dixon JS, Kuntz ID and Venkataraghavan R J. Med. Chem. 29 1986
Docking Small-Molecule Ligands into Active Sites Jones G and Willett P Curr. Opin. Biotechnol. 6 1995
Structure-based Strategies for Drug Design and Discovery Kuntz ID Science 257 1992
Ligand-Protein Docking and Rational Drug Design Lybrand TP Curr. Opin. Struct. Biol. 5 1995
Flexible Docking and Design Rosenfeld R, Vajda S and DeLisi C Annu. Rev. Biophys. Biomol. Struct. 24 1995
A Review of Protein-Small Molecule Docking Methods Taylor RD, Jewsbury PJ and Essex JW J. Comput. Aided. Mol. Des. 16 2002
Docking Treatments¶
Docking treatments are very useful for assessing the quality of candidate prototypes. If the ligand is a known active molecule, docking simulations allow us to identify its bioactive conformation. If the ligand is a candidate molecule, the docking allows us to analyze how it fits to the receptor. The best-docked compounds can be used as leads for further design and optimization.
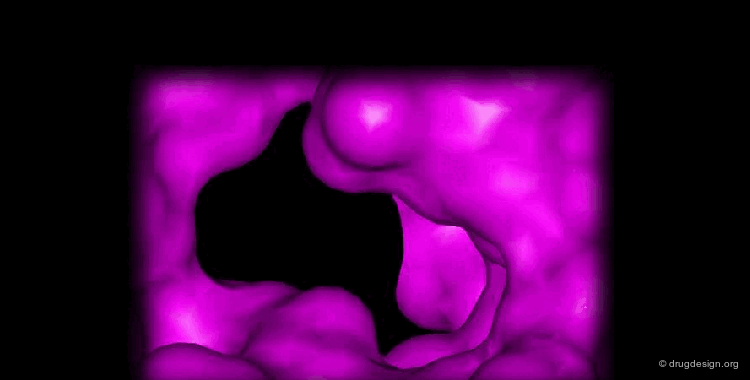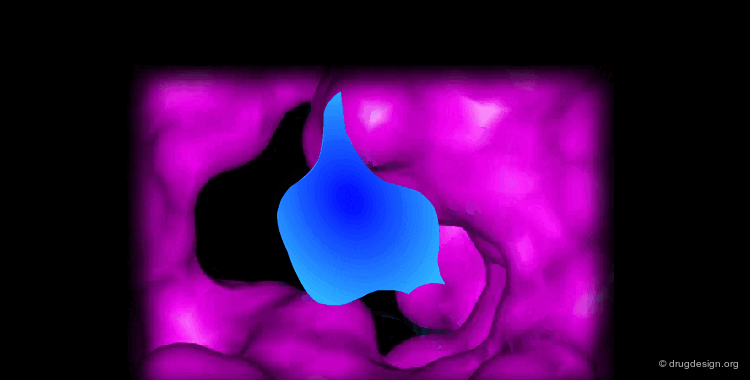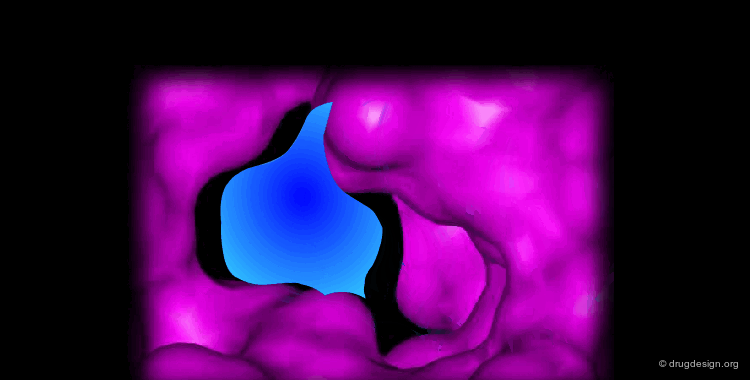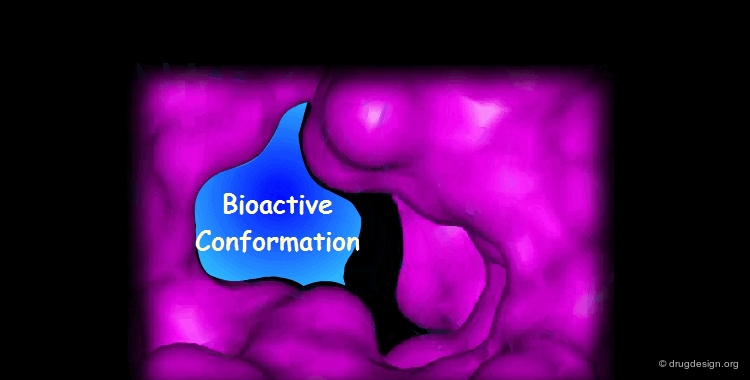
Eight Golden Rules¶
Eight Golden Rules in Receptor-Based Ligand Design¶
Binding efficiency is not only a matter of geometry; the energetics of interactions must be considered. Filling the space in the active site of a protein is a delicate operation. The following examples illustrate some of the important considerations for receptor-based ligand design. Eight rules are presented here.

Rule 1: Coordinate to Key Anchoring Sites¶
When working with target proteins, first one has to consider the proper anchorage of the ligand to key elements of the catalytic site. For example the Zinc of a metalloprotein, the Heme of a cytochrome P450 enzyme, the aspartates of an aspartic protease, the oxyanion hole of serine protease.

Desolvation Upon Binding¶
This anchorage not only positions the ligand in the active site but also counteracts the effect of de-solvating the two components when binding occurs. This is very important energetically.

Rule 2: Exploit Hydrophobic Interactions¶
With hydrophobic pockets, placing a hydrophobic surface of the ligand in hydrophobic sites of the target protein provides an important driving force in complex formation because it reduces non-polar surface areas exposed to water. The more water molecules liberated in drug binding, the greater the gain in entropy and therefore the greater the affinity of the drug for the protein.
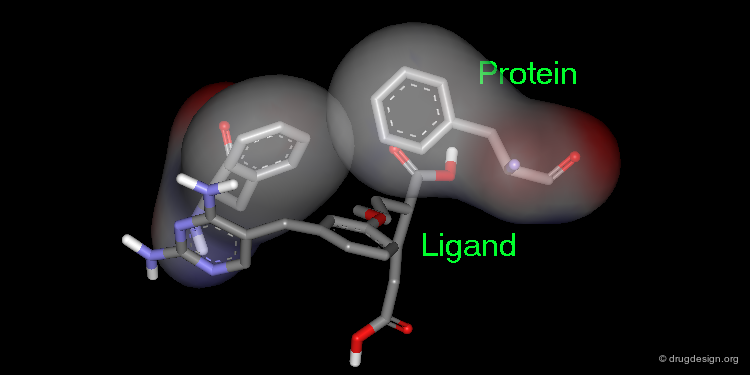
Many Small Contributions¶
Although individually small, the total contribution of hydrophobic bonds to drug-receptor interactions is substantial. Empirical data suggests that the free energy contribution due to hydrophobic bonding is approximately 2.9 kJ/mol per methylene group and 8.4 kJ/mol for a benzene ring. Unlike hydrogen bonds, the hydrophobic interactions are not directional.
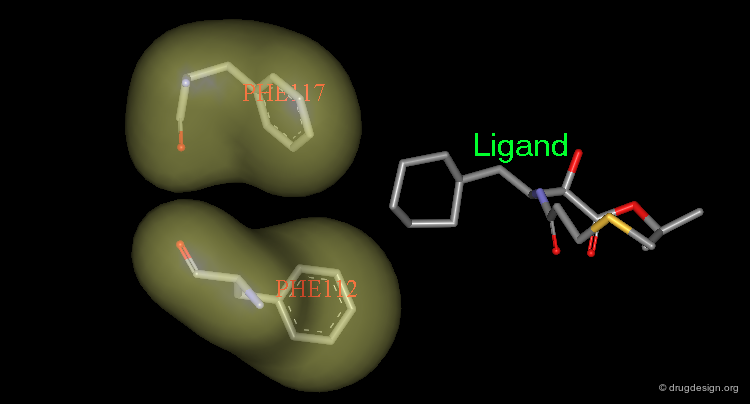
Rule 3: Exploit Hydrogen Bonding Capabilities¶
Once a new skeleton fills a large portion of the available space, a hydrogen-bonding functionality can be placed to complement those functional groups presented either by the protein or water molecules.

Hydrogen Bonds with Backbone Atoms¶
A carbonyl oxygen is optimally satisfied when it accepts two different hydrogen bonds with C=O --- H angles close to 120°. However hydrogen bonds to carbonyl oxygen atoms with a C=O --- H angle close to 180° form the basis for β-sheet formation and are quite favorable.
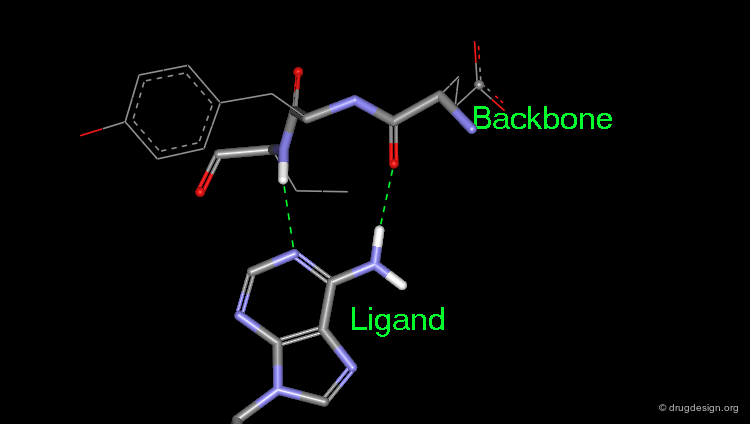
Geometry of a Hydrogen Bond and Solvation Issues¶
The average N-H --- O angle is about 155°, with 90% lying between 140° and 180°. Almost all protein groups are capable of forming hydrogen bonds like this. Where groups are not explicitly hydrogen bonded, they are probably solvated.

Hydrogen Bonds with Residue Atoms¶
Unsatisfied hydrogen bond donors and acceptors are rarely seen in proteins. A corollary for protein-ligand complexes would be burying potential donors and acceptors without forming a complementary hydrogen bond, this would also be highly unfavorable.

Rule 4: Exploit Electrostatic Interactions¶
The optimization of ligand-protein electrostatics can be achieved by placing a positive charge in close vicinity to an enzyme negative charge. The interaction should be fine tuned by subtle structural modifications.

Rule 5: Favor Bioactive Form & Avoid Energy Strain¶
Conformational energy calculations are performed on each design idea in order to determine the internal penalty required for the new ligand to attain its bioactive binding conformation inside the protein. The internal energy that is required for the small molecule to reach its binding conformation is energy lost in binding.

Advantage and Limitation of a Rigid Ligands¶
Restricting the conformation space of an inhibitor can be beneficial to binding when the conformation is biased towards the bioactive conformer. However, if parts of the constricting group generate unfavorable interactions with the protein or induce a non-bioactive conformer, then deleterious effects on binding occur.

Rule 6: Optimize VDW Contacts and Avoid Bumps¶
Attractive van der Waals interactions occur over a short distance range and attraction decreases as 1/r6. As a result, optimization of attractive van der Waals interactions occurs as the shape of the protein binding site and the shape of the ligand match well. A difference of binding energy of 6.3 kJ/mol corresponds to a 10 fold difference in the biological activities.

The Frontier between an Excellent Fit and a Bump¶
The frontier between an excellent fit and a bump is sometimes difficult to recognize visually; it has to be assessed with additional calculations or determined experimentally. Note that calculations of steric fit are difficult because of possible flexing motions of the protein backbone and especially the residue side-chains.

Rule 7: Structural Water Molecules and Solvation¶
Inhibitor design strategies have great potential when they target the displacement of water molecules tightly bound to the protein by incorporating elements of the water molecule within the inhibitor.

Desolvation Energies¶
Desolvation energies are the "price to be paid" for the ligand to attain its binding conformation inside the protein.
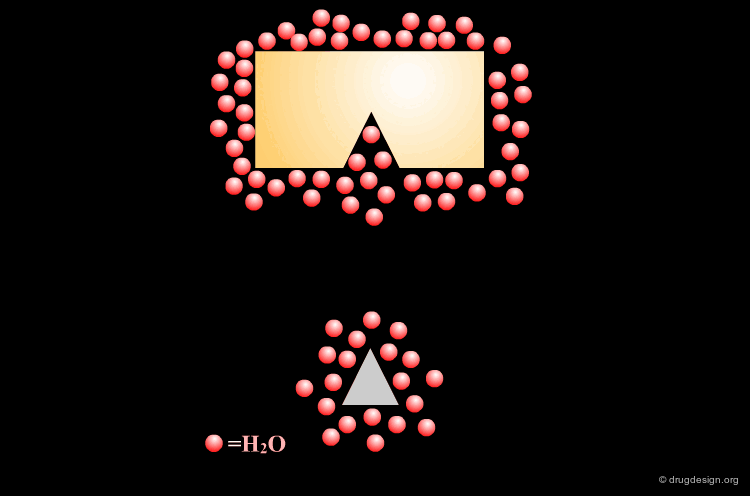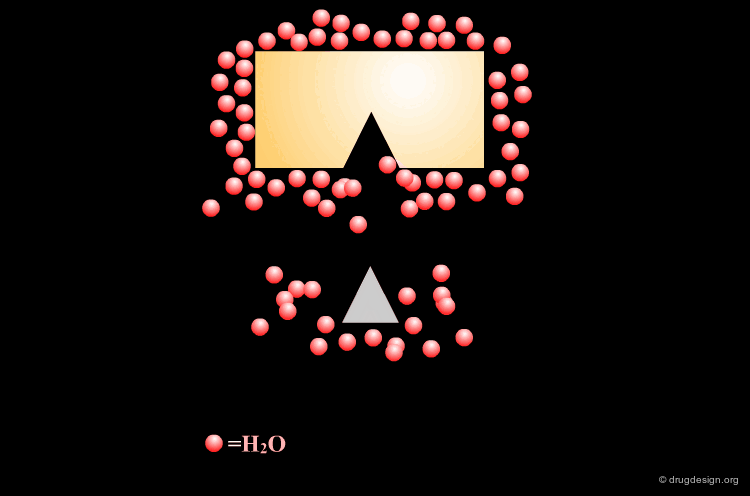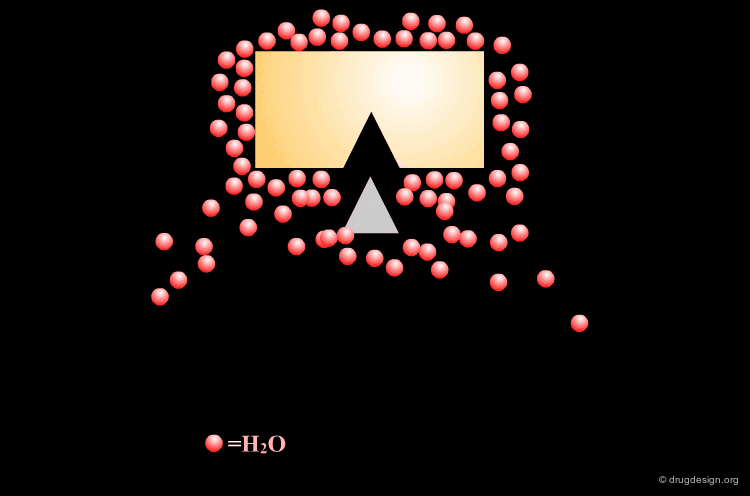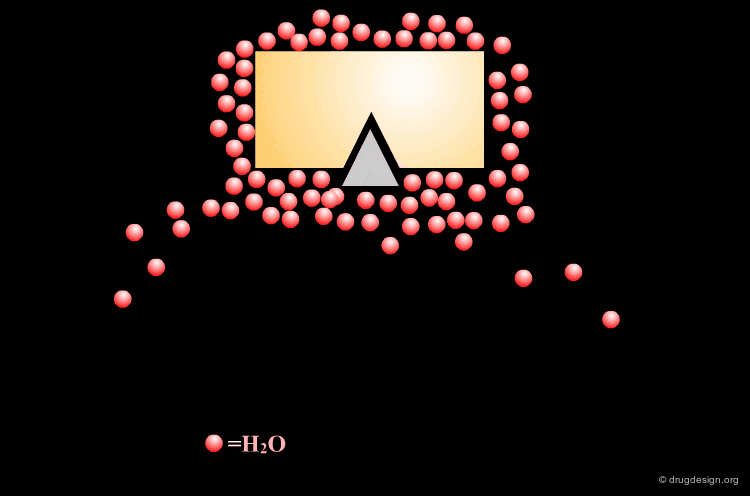
Leave Some Room to Solvate Charged Centers¶
When polar charged groups are considered in the design of a ligand, one should leave some room for other water molecules to solvate the charged center (except possibly when a salt bridge is formed).

Rule 8: Consider Entropic Effect¶
A flexible molecule has a better chance of finding an optimal fit into a receptor, but this is achieved at the cost of large conformational entropy. Sufficient conformational rigidity is essential to ensure that the loss of entropy upon ligand binding is acceptable. A rigid molecule has little conformational entropy but is unlikely to fit optimally into the receptor.
Gaining Binding by Reduction of Entropy¶
Conformational flexibility and the associated free energy of the ligand play an important role in ligand-receptor interactions. An analysis of the contributions of various functional groups to binding demonstrates that each freely rotating bond in a ligand reduces binding free energy by about 2.9 kJ/mol.
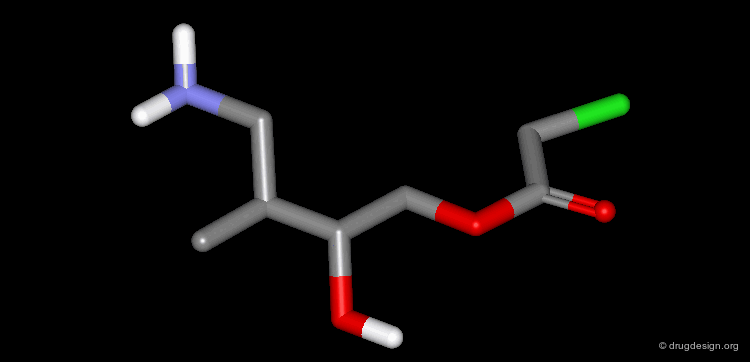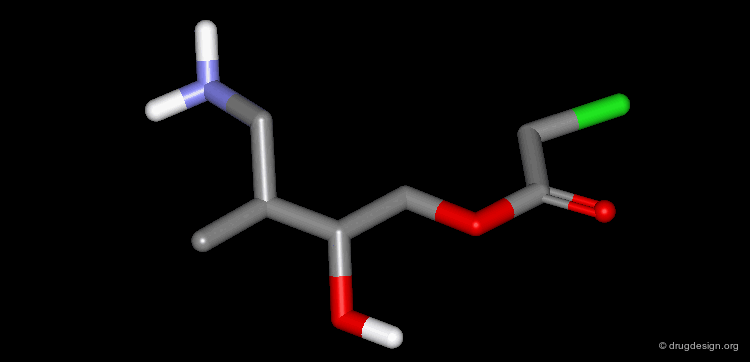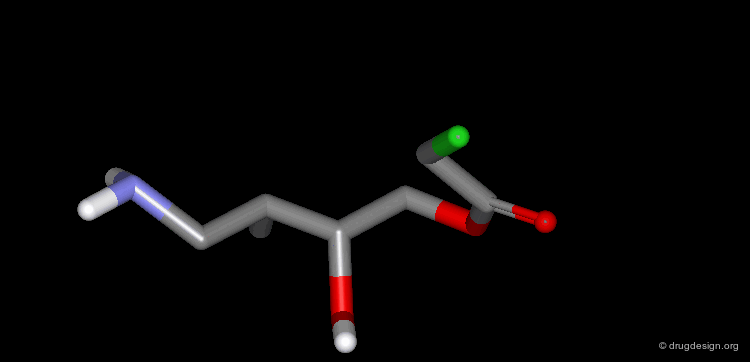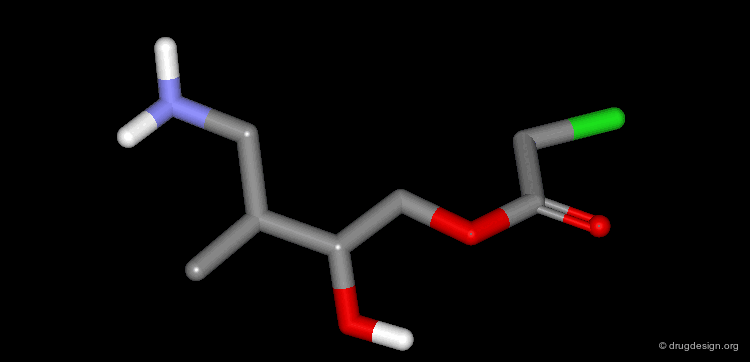
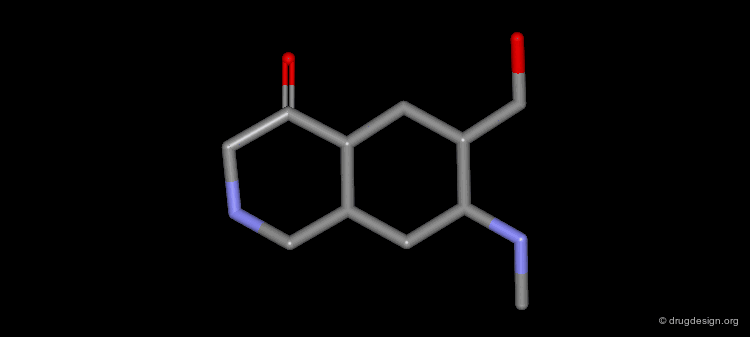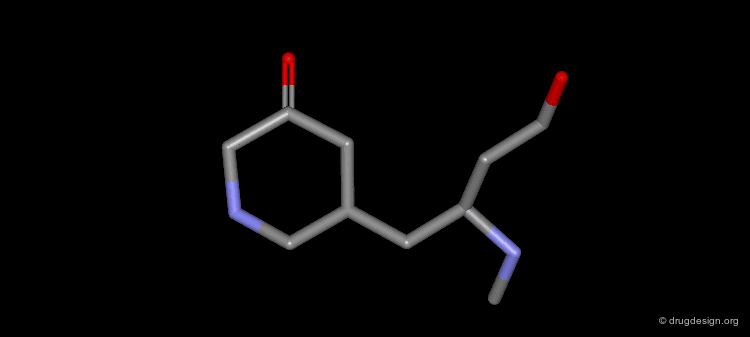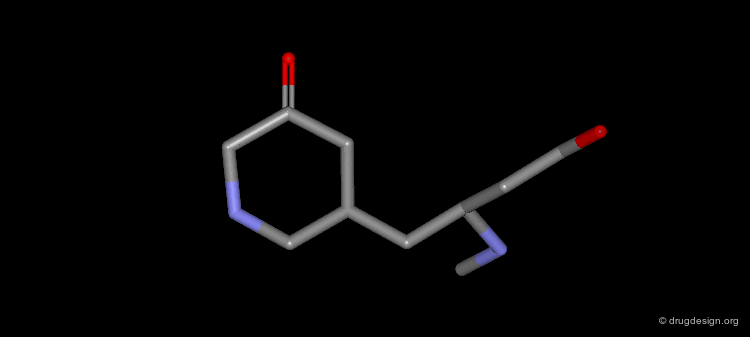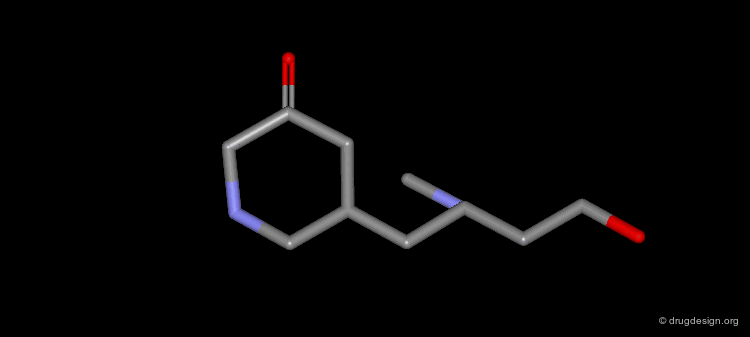
Example of Ligand Rigidification¶
Making a ligand more rigid will lead to enhanced activity if the right conformation is maintained. For example freezing of two rotational degrees of freedom in flexible antiulcer agents in a rigid analog resulted in a 150-fold enhancement in activity.

Making a Flexible Molecule More Rigid¶
Making a flexible molecule more rigid will lead to enhanced activity if the right conformation is maintained.
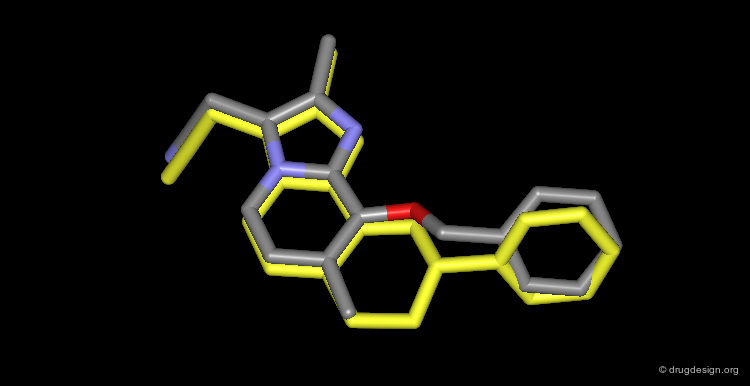
The Four Design Methods¶
The Four Design Methods¶
There are four basic methods in receptor-based drug design: analog design, database searching, computerized de novo design and manual design.

Analog Design¶
Principles of Analog Design¶
In analog design, chemically related molecules are synthesized and tested in order to collect useful data on the structural requirements for the biological activities. In structure-based drug design the structure-activity relationships (SAR) are analyzed in 3D; for example a loss in potency is interpreted as a loss of good complementary between the ligand and the receptor.

Example of Analog Design¶
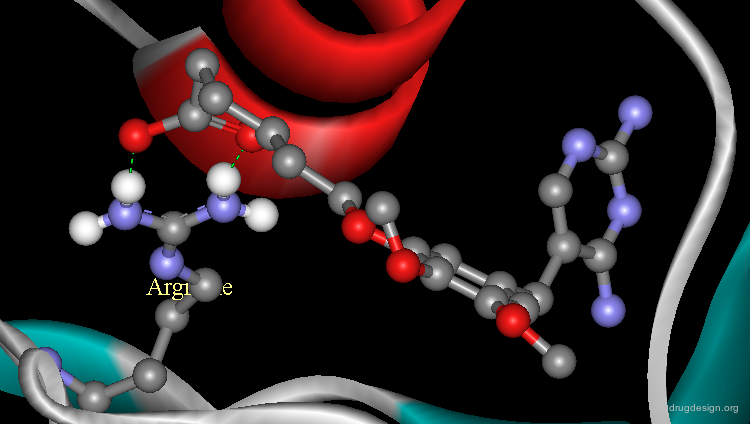
The analysis of the crystal structure of the complex between trimethoprim and dihydrofolate reductase (DHFR) reveals the existence of possible additional binding sites. In particular, a basic arginine residue could be considered for increasing the binding.

articles
Receptor-Based Design of Dihydrofolate Reductase Inhibitors: Comparison of Crystallographically Determined Enzyme Binding With Enzyme Affinity in a Series of Carboxy-Substituted Trimethoprim Analogues Kuyper LF, Roth B, Baccanari DP, Ferone R, Beddell CR, Champness JN, Stammers DK, Dann JG, Norrington FE, Baker DJ and Goodford PJ J. Med. Chem. 28 1985
Crystal structures of Escherichia coli and Lactobacillus casei dihydrofolate reductase refined at 1.7 A resolution. I. General features and binding of methotrexate Bolin JT, Filman DJ, Matthews DA, Hamlin RC, Kraut J J. Biol. Chem. 257 1982
Additional Binding with Arginine Residue¶
The analysis indicated that a carboxyl function introduced in the chemical structure of trimethoprim could form an ionic linkage with the arginine residue. The carboxyl analog proved to be significantly more potent than the reference trimethoprim and its X-ray structure confirmed the validity of this finding.
Database Searching¶
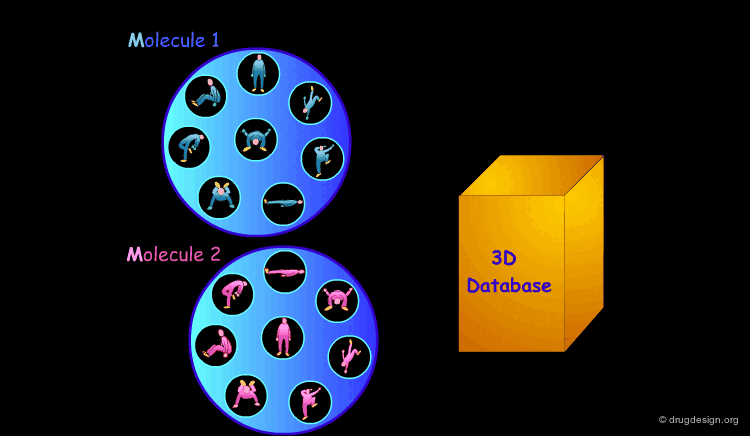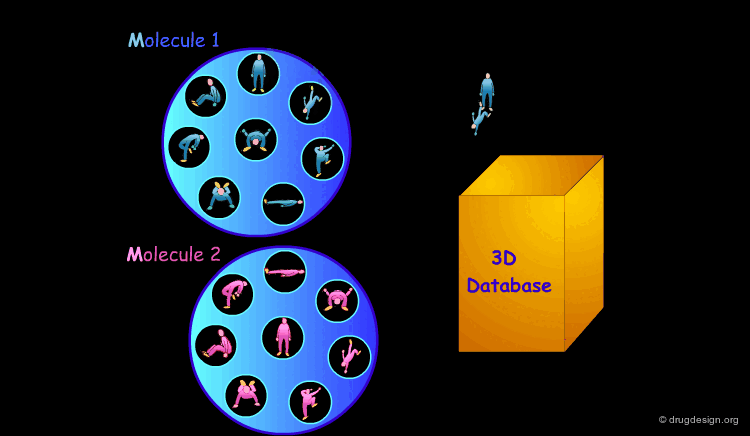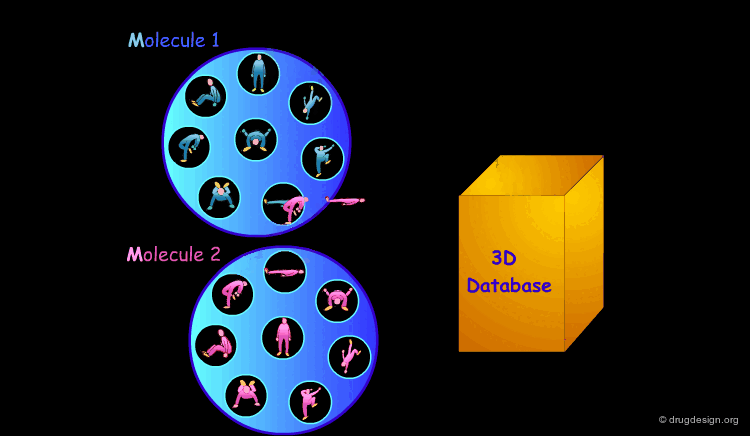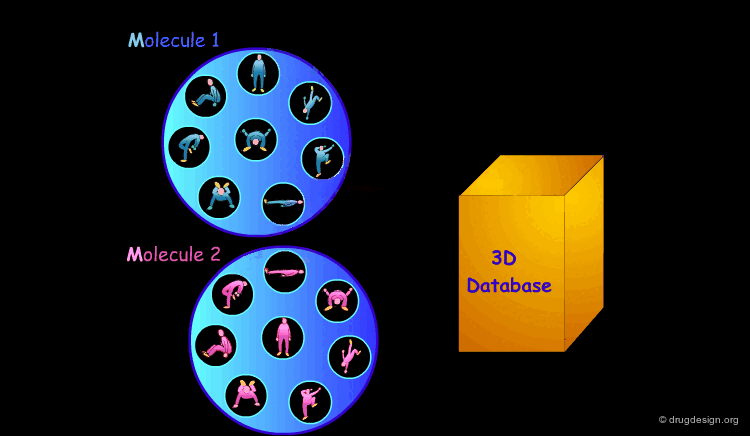
3D Database Searching¶
It is sometimes a challenge to design de novo leads on the basis of an unliganded protein structure alone. In practice the database approach provides an easy way to arrive at leads. It consists of a 3D computerized search of a database of synthesized or naturally occurring compounds and testing them in silico. In this process each candidate molecule is automatically inserted, oriented and minimized into the active site.

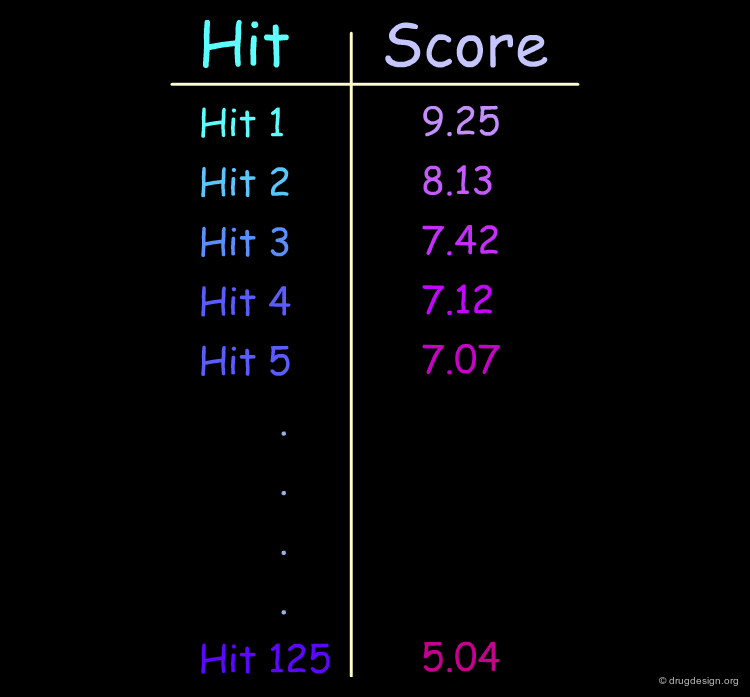
Scoring the Hits¶
The result of this treatment is a list of compounds ranked by a score based on the overall match with the receptor.
Advantages of Database Searching¶
An advantage of this method is that it generally does not require special synthetic efforts (the molecule is available) but on the other hand it is highly dependent on the content of the database and also it is time-consuming.

Problems of Conformational Complexity¶
Due to the conformational complexity of the current molecules, only a few of their conformations are actually stored in the databank. Unless complicated conformational generators are incorporated in the calculations, database searching cannot be exhaustive and therefore many molecules may be overlooked in the process.
articles
Pharmacophoric Pattern Matching in Files of Three-Dimensional Chemical Structures: Comparison of Conformational-Searching Algorithms for Flexible Searching Clark DE, Jones G, Willett P, Kenny PW, and Glen RC J. Chem. Inf. Comput. Sci. 34 1994
Rapid Calculation of Coordinates from Distance Matrices Crippen GM J. Comput. Phys. 26 1978
Use of Flexible Queries for Searching Conformationally Flexible Molecules in Databases of Three-Dimensional Structures Guner OF, Henry DR and Pearlman RS J. Chem. Inf. Comput. Sci. 32 1992
Three-Dimensional Shape-Based Searching of Conformationally Flexible Compounds Hahn MA J. Chem. Inf. Comput. Sci. 37 1997
Flexible 3D Searching: The Directed Tweak Technique Hurst T J. Chem. Inf. Comput. Sci. 34 1994
Distance Geometry Kuntz ID, Thomason JF, Oshiro CM Methods Enzymol. 177 1989
book
Crippen GM Distance Geometry and Conformational Calculations Research Studies Press, Wiley 1981
Crippen GM and Havel TF Distance Geometry and Molecular Conformation Wiley 1988
Assessing the Validity of the 3D Structures¶
Whenever results are obtained from a database search, one should always be critical and check the validity of the result, especially its geometry. For example, using a query consisting of 4 phenyl rings and one OH group, the following search result was obtained. Although geometrically correct, the conformation of the result with all the substituents in the axial orientation is highly unrealistic (too high energy).

Example of Database Searching¶
In order to identify small molecules that are complementary in shape to the active site of HIV-1 protease, the 3D structure of the enzyme was used as a screen. The following benzophenone derivative was found as an HIV-1 inhibitor (IC50 = 11 mM).

Limitations in Database Approaches¶
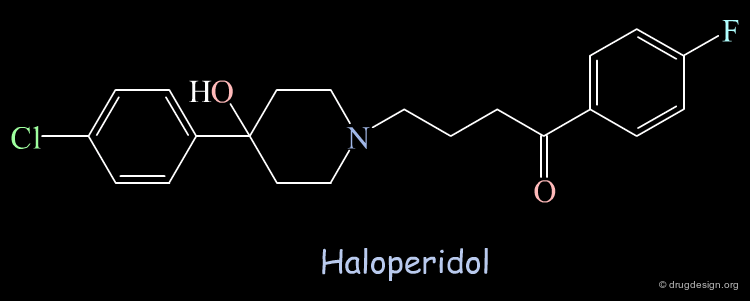
Using the crystallographic structure of the HIV-1 protease for screening, Haloperidol was found as a "hit" able to bind to the catalytic site. The molecule was tested and showed moderate inhibitory activity (Ki = 100 µM).
articles
Structure-Based Design of Nonpeptide Inhibitors Specific for the Human Immunodeficiency Virus 1 Protease DesJarlais RL, Seibel GL, Kuntz ID, Furth PS, Alvarez JC, Ortiz de Montellano PR, DeCamp DL, Babe LM and Craik CS Proc. Natl. Acad. Sci. 87 1990
Unexpected Binding Mode of Haloperidol¶
Subsequent X-ray crystallographic studies revealed that the structure was not binding in the catalytic site as foreseen initially, but in another region 5 Å away and rotated by 80° from the orientation predicted in the computational search; the Haloperidol analog is represented here in yellow. It was expected to interact in the region occupied by known inhibitors, as for example MVT-101 represented in pink.

Databases of Molecules in 3D¶
Throughout the world there are thousands of chemical suppliers offering more than 3 million of organic molecules. The advantage of using commercially available molecules in database searching is that they offer a great diversity and can be purchased and tested rapidly. The 3D structures of molecules are either obtained experimentally (X-ray data, NMR) or by modeling techniques.

The Main Purpose of a 3D-Database Search¶
The main purpose of a 3D-database search is not to find compounds with ultrapotent activities. A realistic goal of a 3D-database search should be the discovery of active search results, which can subsequently be used as the basis for further synthetic modifications.

De-Novo Design¶
Automated Construction Approaches¶
The purpose of construction programs is to discover a new chemical framework that fits to the active site of the target receptor or enzyme. Some methods are based on an existing moiety and additional fragments are appended by a step-by-step build up procedure. Other methods consist of assembling novel molecules from pieces that are positioned optimally in favorable regions of the active site.

articles
The Computer Program LUDI: a New Method for the De-Novo Design of Enzyme Inhibitor Leads Bohm H-J J. Comput. Aided Mol. Des. 6 1992
LUDI: Rule-Based Automatic Design of New Substituents for Enzyme Inhibitor Leads Bohm H-J J. Comput. Aided Mol. Des. 6 1992
Automated Site-Directed Drug Design: the Generation of a Basic Set of Fragments to be Used for Automated Structure Assembly Chau PL and Dean PM J. Comput. Aided Mol. Des. 6 1992
PRO-LIGAND: an Approach to De Novo Molecular Design. 1. Application to the Design of Organic Molecules Clark DE, Frenkel D, Levy SA, Li J, Murray CW, Robson B, Waszkowycz B and Westhead DR J. Comput. Aided Mol. Des. 9 1995
De Novo Design of Enzyme Iinhibitors by Monte Carlo Ligand Generation Gehlhaar DK, Moerder KE, Zichi D, Sherman CJ, Ogden RC and Freer ST J. Med. Chem. 38 1995
SPROUT, HIPPO and CAESA: Tools for De Novo Structure Generation and Estimation of Synthetic Accessibility Gillet VJ, Myatt G, Zsoldos Z and Johnson P Persp. Drug Discov. Des. 3 1995
CAVEAT: a Program to Facilitate the Design of Organic Molecules Lauri G and Bartlett PA J. Comput. Aided. Mol. Des. 8 1994
Automatic Creation of Drug Candidate Structures Based on Receptor Structure: Starting Point for Artificial Lead Generation Nishibata Y and Itai A Tetrahedron 47 1991
Confirmation of Usefulness of a Structure Construction Program Based on Three-Dimensional Receptor Structure for Rational Lead Generation Nishibata Y, Itai A J. Med. Chem. 36 1993
CONCERTS: Dynamic Connection of Fragments as an Approach to De Novo Ligand Design Pearlman DA and Murcko MA J. Med .Chem. 39 1996
A Genetic Algorithm for Structure-Based De Novo Design Pegg SC, Haresco JJ, Kuntz ID J. Comput. Aided Mol. Des. 15 2001
BUILDER v.2: Improving the Chemistry of a De-Novo Design Strategy Roe DC and Kuntz ID J. Chem. Inf. Comput. Sci. 9 1995
GroupBuild: a Fragment-Based Method for De-Novo Drug Design Rotstein SH, Murcko MA J. Med. Chem. 36 1993
Molecule Generated by an Automated Method¶
The crystallographic structure of the immunophillin FKBP-12 was used as input for the design of new ligands, based on a construction approach. Among other solutions, the following structure was proposed. The molecule was synthesized and the biological tests confirmed that the compound was active (Ki = 16 µM).

articles
Design, Synthesis and X-ray Crystallographic Studies of Novel FKBP-12 Ligands Babine RE, Bleckman TM, Kissinger CR, Showalter R, Pelletier LA, Lewis C, Tucker K, Moomaw E, Parge HE and Villafranca JE Bioorg. Med. Chem. Lett. 5 1995
Manual Design¶
Manual Design¶
It is a major challenge to design de novo leads on the basis of an unliganded protein structure alone. However manual design is greatly facilitated by starting from an existing "solution" which consists of the visualization of a complex (an experimental result or a model). This procedure is currently undertaken either by the medicinal chemist or the molecular modeler.

Importance of Visualization¶
In manual design the visualization is of central importance. It helps to see in a condensed way the molecular objects. In addition, it triggers the synergy between knowledge and intuition for identifying key features of the receptor pocket or for building molecules under given requirements.

Tools in Manual Design¶
Real time rotations, molecular editing functions such as deletion, addition, fusion, connections, torsion angle alterations, superimpositions, coloring and stereo constitute the minimum necessary for sketching and assessing the validity of a solution considered in manual design. On a higher level, the energy calculations and minimizations can complete the panoply of the synthetic chemist.

Fully Exploiting the Fruits of the Analyses¶
Manual design aims at the rapid exploitation of the information revealed by molecules previously tested and the translation of this knowledge into the structure of a new molecule to be synthesized. In this example, the analyses of known active molecules indicated the existence of two hydrogen bonds as indicated in the view. However only one hydrogen bond is exploited by the known molecules.

Design of a Hybrid Molecule¶
The designed keto molecule incorporates knowledge extracted from the two other active phenyl-imidazole analogs. The imidazole ring of each of the compounds is mimicked by a pyridine ring in the designed structure. The resulting compound proved to be much more potent than the other two compounds.

Another Iteration¶
Another Round of Analysis & Design¶
The new biological results, the availability of a new X-ray structure, the improved understanding of structure-activity relationships, the optimization of a series, and the need for an entirely new chemical structure are all reasons for the medicinal chemist to return to the "analysis" or "design" modes. The information is used as the basis for another round of analysis, design, synthesis and compound testing.

A Success Story¶
Example of Successful Structure-Based Design¶
The use of the crystallographic structure of the HIV-1 protease in drug design represents one of the more impressive success stories in the structure-based drug design field. Based on the structure of the protein, many groups have used an iterative structure-based design process. This has resulted in the identification of distinct classes of inhibitors and several successful drug candidates have emerged from these studies and are used in the control of AIDS.

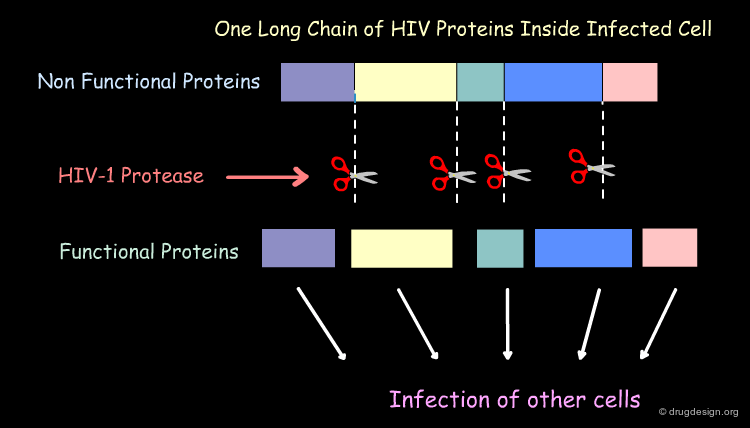
Mechanism of Action of the HIV-1 Protease¶
The HIV-1 protease plays a crucial part in the life cycle of the HIV virus. The task of the HIV-1 protease is to cut this long 'polyprotein' into the proper protein-sized pieces. As a result of this crucial task, HIV-1 protease is an excellent target for drug therapy. Inhibitor drugs block the action of the protease and the virus perishes because it is unable to mature into its infectious form.
The Crystallographic Structure of the HIV-1 Protease¶
The HIV-1 protease is a small dimer enzyme comprising two identical folded 99 amino-acid chains A and B. This view represents the α carbons of the HIV-1 protease.

articles
Three-Dimensional Structure of Aspartyl Protease from Human Immunodeficiency Virus HIV-1 Navia MA, Fitzgerald PM, McKeever BM, Leu CT, Heimbach JC, Herber WK, Sigal IS, Darke PL and Springer JP Nature 337 1989
Structure of Complex of Synthetic HIV-1 Protease with a Substrate Inhibitor at 2.3 A Resolution Miller M, Schneider J, Sathyanarayana BK, Toth MV, Marshall GR, Clawson L, Selk L, Kent SB and Wlodawer A Science 246 1989
Hydrophobic Cavity of the HIV-1 Protease¶
The assembly of the two chains of the HIV-1 protease forms a hydrophobic cavity at the center of the catalytic site of the enzyme.

Flap of the HIV-1 Protease¶
An interesting feature of the structure is the "flap region" in each chain that arches over the active site. Relative to the free enzyme, the complex exhibits backbone movements as the flap regions close jaw-like over the substrate.

Transition State Concept for the Design of Inhibitors¶
Since the key catalytic step for virus replication is breaking the peptide bond, analogues were designed which resemble the transition state intermediate but are not cleaved by the enzyme.

Topography of the Active Site of the Enzyme¶
It was found that several hexapeptides were inhibitors of HIV-1 protease. Based on these results, it was suggested that the first requirement is that the inhibitor must have side chains which will enter in 6 subpockets (P1, P2, P3, P1', P2', P3') of the enzyme.

The MVT-101 Inhibitor¶
The MVT-101 compound was found to inhibit the HIV-1 protease (Ki = 780 nM). It is a hexapeptide in which the scissile peptide bond (that which is cleaved by the protease) is replaced by an aminomethylene group.

articles
Structure of complex of synthetic HIV-1 protease with a substrate-based inhibitor at 2.3 A resolution Miller M, Schneider J, Sathyanarayana BK, Toth MV, Marshall GR, Clawson L, Selk L, Kent SB and Wlodawer A Science 246 1989
Crystallographic Resolution of the HIV-1 Protease¶
A major milestone in HIV-1 inhibition occurred in 1989 with the crystallographic determination of the three dimensional structure of the enzyme. A decade later, more than four hundred structures were available in the PDB, including several genetic strains of the enzyme, complexes of the enzyme with many different drugs and inhibitors, and dozens of mutant enzymes. This information played a major role in the successful development of drugs against AIDS.

articles
Three-Dimensional Structure of Aspartyl Protease from Human Immunodeficiency Virus HIV-1 Navia MA, Fitzgerald PM, McKeever BM, Leu CT, Heimbach JC, Herber WK, Sigal IS, Darke PL and Springer JP Nature 337 1989
Structure of Complex of Synthetic HIV-1 Protease with a Substrate Inhibitor at 2.3 A Resolution Miller M, Schneider J, Sathyanarayana BK, Toth MV, Marshall GR, Clawson L, Selk L, Kent SB and Wlodawer A Science 246 1989
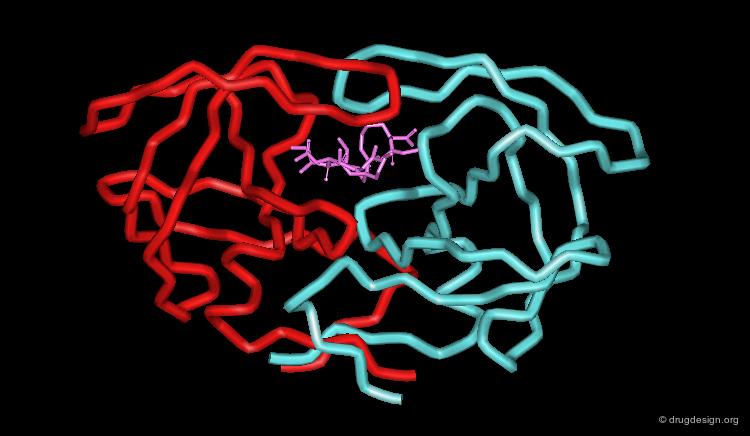
X-ray of the Complex of MVT-101 with the Enzyme (1/3)¶
The complex of MVT-101 with the HIV-1 protease is one of the first crystallographic complexes that was solved. It revealed the binding mode of MVT-101 that proved to be of great importance for the discovery of effective drugs.
articles
Structure of Complex of Synthetic HIV-1 Protease with a Substrate Inhibitor at 2.3 A Resolution Miller M, Schneider J, Sathyanarayana BK, Toth MV, Marshall GR, Clawson L, Selk L, Kent SB and Wlodawer A Science 246 1989
X-ray of the Complex of MVT-101 with the Enzyme (2/3)¶
A key feature of HIV-1 protease inhibitors is the hydrogen bonding formed with the two central carbonyls to a water molecule which acts, via hydrogen bonding, as a bridge to Ile-50 and Ile-50' in the flaps of HIV-1 protease and stabilizes the ''closed" conformation.

articles
Structure of Complex of Synthetic HIV-1 Protease with a Substrate Inhibitor at 2.3 A Resolution Miller M, Schneider J, Sathyanarayana BK, Toth MV, Marshall GR, Clawson L, Selk L, Kent SB and Wlodawer A Science 246 1989
X-ray of the Complex of MVT-101 with the Enzyme (3/3)¶
The inhibitors have also a very extended hydrogen bond network with other amino acids in the catalytic site. For example,MVT-101 forms hydrogen bonds with Ile-50, Ile-50', Gly-48, Gly-48', Asp-29, Asp-29', Gly-27 and Gly-27'.

articles
Structure of Complex of Synthetic HIV-1 Protease with a Substrate Inhibitor at 2.3 A Resolution Miller M, Schneider J, Sathyanarayana BK, Toth MV, Marshall GR, Clawson L, Selk L, Kent SB and Wlodawer A Science 246 1989
Design of Peptide-Like Structures¶
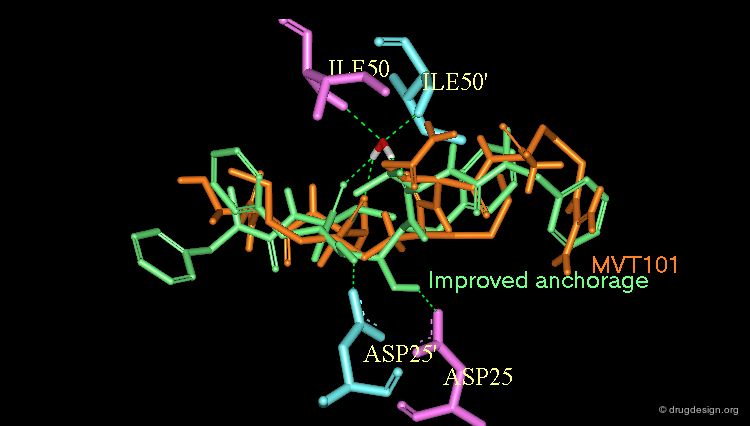
The information provided by the X-ray complex of MVT-101 with the protease gave the impetus to the discovery of potent peptidomimetic molecules. Peptide-like structures were designed to anchor not only to Ile-50 and Ile-50' but also to Asp-25 and Asp-25' , that stabilize the "closed" conformation of the enzyme.
articles
Influence of Stereochemistry on Activity and Binding Modes for C2 Symmetry-Based Diol Inhibitors of HIV-1 Protease Hosur M, Bhat T, Kempf D, Baldwin E, Liu B, Gulnik S, Wideburg N, Norbeck D, Apelt K and Erickson J J. Am. Chem. Soc. 116 1994
Symmetry-based Inhibitors of HIV Protease. Structure-activity Studies of Acylated 2,4-Diamino-1,5-Diphenyl-3-Hydroxypentane and 2,5-Diamino-1,6-Diphenylhexane-3,4-Diol Kempf DJ, Codacovi L, Wang XC, Kohlbrenner WE, Wideburg NE, Saldivar A, Vasaanonda S, Marsh CK, Bryant P, Sham HL, Green BE, Betebenner DA, Erickson J and Norbeck DW J. Med. Chem. 36 1993
Progress in the Discovery of Orally Bioavailable Inhibitors of HIV Protease Kempf DJ Perspect. Drug. Discov. Design.
1994
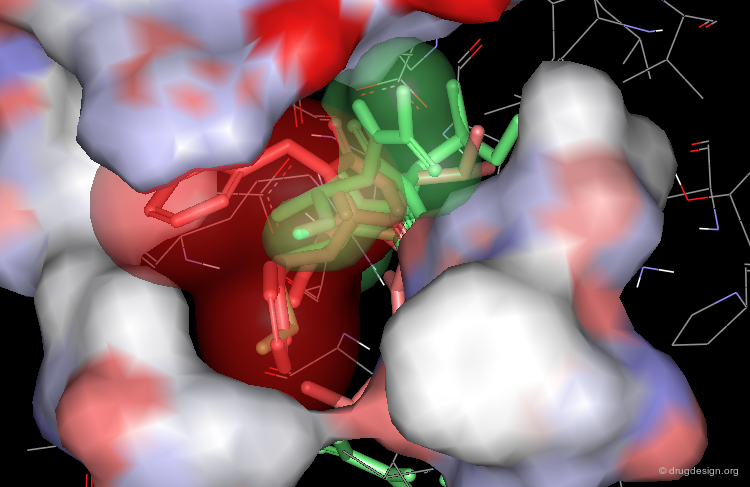
Optimization to Fit the hydrophobic pockets¶
Additional efforts were also made to better fill the hydrophobic pockets (green=MVT-101, red=designed).
Example of Optimized Structure¶
A typical example of a peptidic compound that bears these changes is A-77003, which is about 5000 times more potent than MVT-101 (pKi = 0.15 nM).

X-ray Structure of the A-77003 Complex (1/4)¶
Analysis of the complex shows that A-77003 (in which the scissile amide bond of the substrate is replaced by a diol moiety) anchors well on the two aspartic acids Asp-25 and Asp-25'.

X-ray Structure of the A-77003 Complex (2/4)¶
A-77003 also binds to Ile-50 and Ile-50' through a water molecule.

X-ray Structure of the A-77003 Complex (3/4)¶
The hydrophobic pockets are well filled by phenyl and isopropyl groups.

X-ray Structure of the A-77003 Complex (4/4)¶
The inhibitor forms several additional hydrogen bonds with the enzyme.

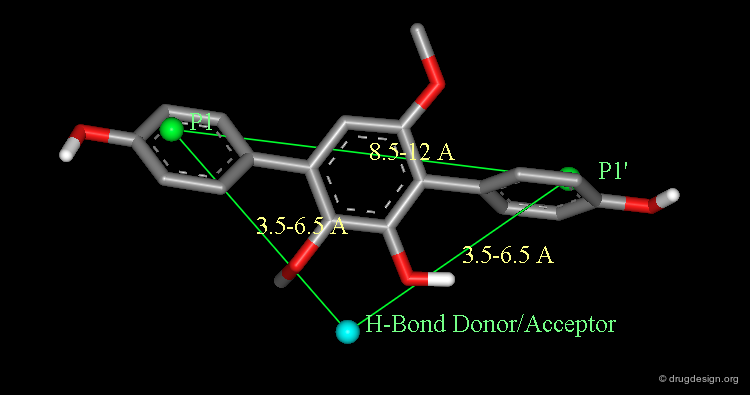
A Drug Design Solution Using Database Searching¶
In order to improve the bioavailability of the inhibitors a data-base approach was considered. Based on the docked A77003 diol/HIV-1 protease complex, a simple pharmacophore was defined by the intramolecular distances between the symmetric hydrophobic groups (P1 and P1') and a hydrogen bond donor/acceptor group bound to the catalytic aspartates.

articles
Cyclic HIV Protease Inhibitors Capable of Displacing the Active Site Structural Water Molecule De Lucca VG, Erickson-Viitanen S and Lam PYS Drug Discovery Today 2 1997
Rational Design of Potent, Bioavailable, Nonpeptide Cyclic Ureas as HIV Protease Inhibitors Lam PYS, Jadhav PK, Eyermann CJ, Hodge CN, Ru Y, Bacheler LT, Meek JL, Otto MJ, Rayner MM, Wong YN, Chang C-H, Weber PC, Jacson DA, Sharpe TR and Erickson-Viitanen S Science 263 1994
The Terphenyl Hit¶
Using this query in a 3D database search resulted in the following terphenyl derivative hit:
articles
Cyclic HIV Protease Inhibitors Capable of Displacing the Active Site Structural Water Molecule De Lucca VG, Erickson-Viitanen S and Lam PYS Drug Discovery Today 2 1997
Rational Design of Potent, Bioavailable, Nonpeptide Cyclic Ureas as HIV Protease Inhibitors Lam PYS, Jadhav PK, Eyermann CJ, Hodge CN, Ru Y, Bacheler LT, Meek JL, Otto MJ, Rayner MM, Wong YN, Chang C-H, Weber PC, Jacson DA, Sharpe TR and Erickson-Viitanen S Science 263 1994
In Depth Analysis of the Hit¶
The following analyses determined the successful continuation of the project; docking experiments of the terphenyl derivative in the HIV-1 protease showed that the top methoxy group is mimicking the structural water whereas the bottom hydroxy and methoxy groups bind to the Asp-25.

articles
Cyclic HIV Protease Inhibitors Capable of Displacing the Active Site Structural Water Molecule De Lucca VG, Erickson-Viitanen S and Lam PYS Drug Discovery Today 2 1997
Rational Design of Potent, Bioavailable, Nonpeptide Cyclic Ureas as HIV Protease Inhibitors Lam PYS, Jadhav PK, Eyermann CJ, Hodge CN, Ru Y, Bacheler LT, Meek JL, Otto MJ, Rayner MM, Wong YN, Chang C-H, Weber PC, Jacson DA, Sharpe TR and Erickson-Viitanen S Science 263 1994
Incorporating Binding Features of the Water Molecule¶
Based on this analysis it was deduced that the incorporation of the binding features of the water molecule into the inhibitor structure would be beneficial because its displacement should be energetically favored.

Rigidification of the Linear Inhibitor¶
Also, a rigidification of the linear inhibitor was predicted to provide a positive entropic effect (less conformations available).

Considerations for Specificity¶
Moreover, mimicking the water molecule within the inhibitor should ensure HIV protease specificity against other aspartic proteases, as only retroviral proteases share this structural water.

Design of Cyclohexanone Scaffold¶
A new scaffold cyclohexanone derivative was then designed based on these concepts.

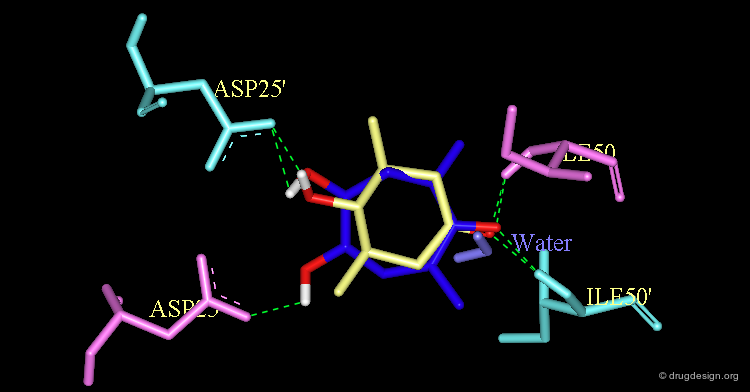
The Design of Cyclic Ureas¶
Modeling studies were performed and showed that replacing the cyclohexanone by a cyclic-urea (7-membered ring) increased the binding to the enzyme. The change provided a better hydrogen bonding to the Asp-25 and Asp-25' as illustrated below.
articles
Cyclic HIV Protease Inhibitors Capable of Displacing the Active Site Structural Water Molecule De Lucca VG, Erickson-Viitanen S and Lam PYS Drug Discovery Today 2 1997
Rational Design of Potent, Bioavailable, Nonpeptide Cyclic Ureas as HIV Protease Inhibitors Lam PYS, Jadhav PK, Eyermann CJ, Hodge CN, Ru Y, Bacheler LT, Meek JL, Otto MJ, Rayner MM, Wong YN, Chang C-H, Weber PC, Jacson DA, Sharpe TR and Erickson-Viitanen S Science 263 1994
Limitation of the 6-Membered Ring Scaffold¶
The 6-membered ring did not provide the proper directions to reach the two other pockets.

The 7-Membered cyclic Urea Scaffold¶
The 7-membered cyclic urea provided the exact stereochemical features to anchor to the aspartates, to mimic the structural water molecule and to provide the correct directions of substitution for accessing to the four hydrophobic pockets.

Design of the Cyclic Urea XK-263¶
All this effort led to the design and modeling of an entirely novel series of potent inhibitors: cyclic urea compounds such as XK-263 (pKi = 0.31 nM).

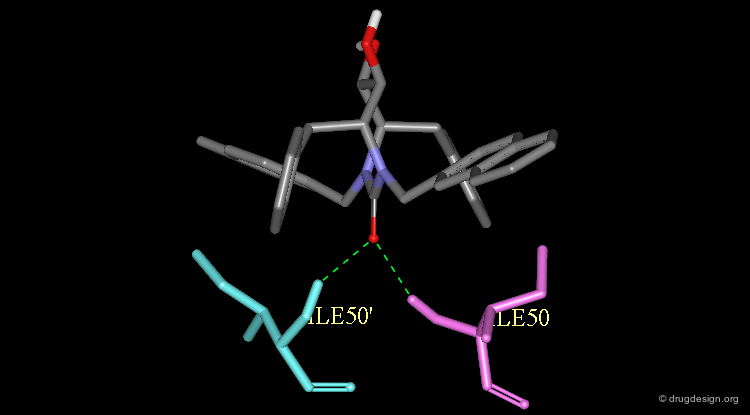
The Crystallographic Structure of XK-263 Complex (1/3)¶
The validity of the design was finally experimentally proven as shown here by the X-ray structure of the complex of XK-263 with the HIV-1 protease. Notice the following features: The carbonyl oxygen mimics the structural water and binds directly to Ile-50 and Ile-50'.
articles
Cyclic HIV Protease Inhibitors Capable of Displacing the Active Site Structural Water Molecule De Lucca VG, Erickson-Viitanen S and Lam PYS Drug Discovery Today 2 1997
Rational Design of Potent, Bioavailable, Nonpeptide Cyclic Ureas as HIV Protease Inhibitors Lam PYS, Jadhav PK, Eyermann CJ, Hodge CN, Ru Y, Bacheler LT, Meek JL, Otto MJ, Rayner MM, Wong YN, Chang C-H, Weber PC, Jacson DA, Sharpe TR and Erickson-Viitanen S Science 263 1994
The Crystallographic Structure of XK-263 Complex (2/3)¶
The diol moiety anchors well to the two aspartic acids Asp-25 and Asp-25'.

articles
Cyclic HIV Protease Inhibitors Capable of Displacing the Active Site Structural Water Molecule De Lucca VG, Erickson-Viitanen S and Lam PYS Drug Discovery Today 2 1997
Rational Design of Potent, Bioavailable, Nonpeptide Cyclic Ureas as HIV Protease Inhibitors Lam PYS, Jadhav PK, Eyermann CJ, Hodge CN, Ru Y, Bacheler LT, Meek JL, Otto MJ, Rayner MM, Wong YN, Chang C-H, Weber PC, Jacson DA, Sharpe TR and Erickson-Viitanen S Science 263 1994
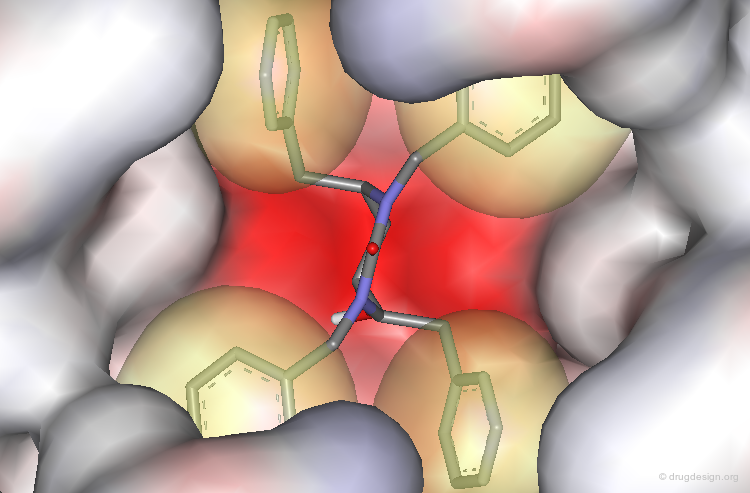
The Crystallographic Structure of XK-263 Complex (3/3)¶
The benzyl and naphthyl groups fill the hydrophobic pockets P1, P1', P2 and P2'.
articles
Cyclic HIV Protease Inhibitors Capable of Displacing the Active Site Structural Water Molecule De Lucca VG, Erickson-Viitanen S and Lam PYS Drug Discovery Today 2 1997
Rational Design of Potent, Bioavailable, Nonpeptide Cyclic Ureas as HIV Protease Inhibitors Lam PYS, Jadhav PK, Eyermann CJ, Hodge CN, Ru Y, Bacheler LT, Meek JL, Otto MJ, Rayner MM, Wong YN, Chang C-H, Weber PC, Jacson DA, Sharpe TR and Erickson-Viitanen S Science 263 1994
Lessons From HIV-1 Protease Inhibition¶
The key learnings acquired in the course of this study are as follows: (1) the project took advantage of previous knowledge of other projects (renin inhibition; renin is an aspartic protease); (2) medicinal chemistry played a pivotal role in bringing data of high informational content at the beginning of the project and in enabling the design of tailor-made structures that were relatively easy to synthesize; (3) X-ray crystallography played a key role in revealing the structural features of the enzyme and the binding modes of the inhibitor. This allowed efficient design of non-peptidic structures and the rapid optimization of the lead compounds; (4) database searching provided an intermediate molecule with information of high informational content. Finally it paved the way for other projects to aim at converting the knowledge of a substrate or a peptide inhibitor into the structure of a non-peptidic drug.

Conclusion¶
Conclusion¶
The molecular recognition of protein-ligand complexes is highly complex and the utilization of an iterative design process has greatly advanced the use of protein structure for the design of drug candidates. The state-of-the-art design process relies, in a large part, on a qualitative understanding of the molecular recognition of protein-ligand complexes based upon analogies to other systems and the use of several methods in parallel.

Copyright © 2024 drugdesign.org