Case Studies in SAR Analyses¶
Info
Typical examples of S.A.R analyses are presented in this chapter to make it easier to understand structure-activity relationships. The examples show how to observe and carefully differentiate structure-activity observations in minute detail and the ways in which the structural determinants behind the biological activities of a series can be recognized.
Number of Pages: 85 (±2 hours read)
Last Modified: January 2009
Prerequisites: Principles of Rational Drug Design, Structure-Based Drug Design: Analysis, Structure-Based Drug Design: Design
Case Study-1 : Banyu Example¶
The Banyu Story with the Urea Structure¶
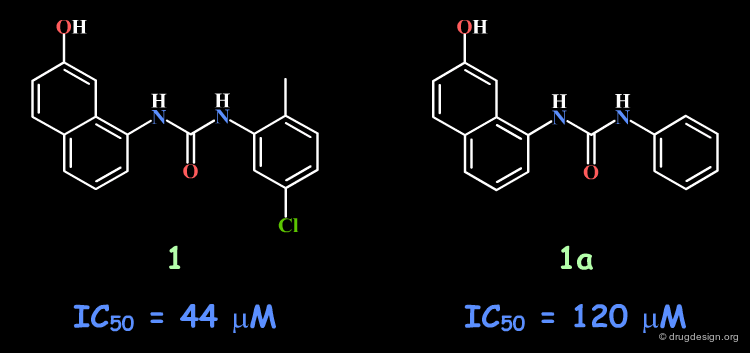
Following a structure-based strategy, the Banyu group identified a hit (Cdk4 inhibitors project) having an urea moiety with an IC50 of 44 µM. The hit was simplified (ortho substituent removed) in order to derive further knowledge based on SAR analyses. In the following pages an outline of the important steps that enabled successful exploitation of SAR is presented.
articles
Structure-Based Generation of a New Class of Potent Cdk4 Inhibitors: New de Novo Design Strategy and Library Design Honma T, Hayashi K, Aoyama T, Hashimoto N, Machida T, Fukasawa K, Iwama T, Ikeura C, Ikuta M, Suzuki-Takahashi I, Iwasawa Y, Hayama T, Nishimura S and Morishima H J. Med. Chem 44 2001
A Novel Approach for the Development of Selective Cdk4 Inhibitors: Library Design Based on Locations of Cdk4 Specific Amino Acid Residues Honma T, Yoshizumi T, Hashimoto N, Hayashi K, Kawanishi N, Fukasawa K, Takaki T,Ikeura C, Ikuta M, Suzuki-Takahashi I, Hayama T, Nishimura S and Morishima H J. Med. Chem 44 2001
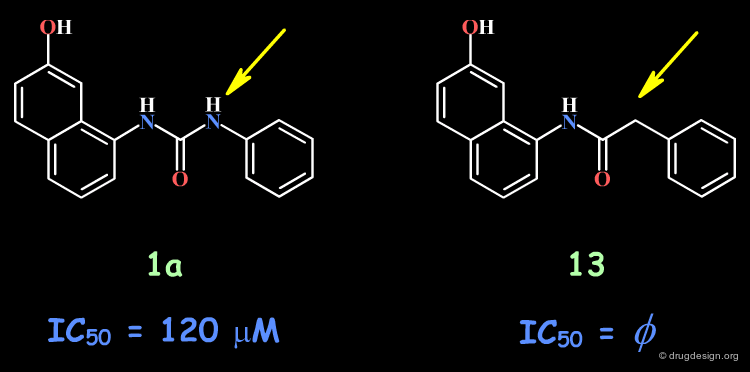
Importance of the Entire Urea Moiety¶
The complete loss of biological activity observed for the analog in which the anilino nitrogen atom is replaced by a carbon shows that the entire urea moiety is important for biological activities.
articles
Structure-Based Generation of a New Class of Potent Cdk4 Inhibitors: New de Novo Design Strategy and Library Design Honma T, Hayashi K, Aoyama T, Hashimoto N, Machida T, Fukasawa K, Iwama T, Ikeura C, Ikuta M, Suzuki-Takahashi I, Iwasawa Y, Hayama T, Nishimura S and Morishima H J. Med. Chem 44 2001
A Novel Approach for the Development of Selective Cdk4 Inhibitors: Library Design Based on Locations of Cdk4 Specific Amino Acid Residues Honma T, Yoshizumi T, Hashimoto N, Hayashi K, Kawanishi N, Fukasawa K, Takaki T,Ikeura C, Ikuta M, Suzuki-Takahashi I, Hayama T, Nishimura S and Morishima H J. Med. Chem 44 2001
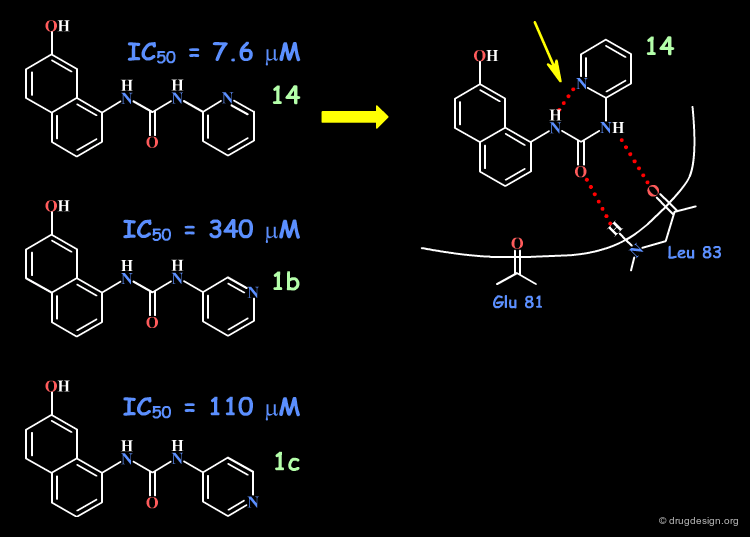
Bioactive Conformation?¶
Based on modeling analyses the Banyu group suspected that the binding of the compounds involved the N-H and the carbonyl by forming two hydrogen bonds with the protein, in a bidentate manner. This hypothesis requires a "cis" conformation for the amide fragment concerned.

articles
Structure-Based Generation of a New Class of Potent Cdk4 Inhibitors: New de Novo Design Strategy and Library Design Honma T, Hayashi K, Aoyama T, Hashimoto N, Machida T, Fukasawa K, Iwama T, Ikeura C, Ikuta M, Suzuki-Takahashi I, Iwasawa Y, Hayama T, Nishimura S and Morishima H J. Med. Chem 44 2001
A Novel Approach for the Development of Selective Cdk4 Inhibitors: Library Design Based on Locations of Cdk4 Specific Amino Acid Residues Honma T, Yoshizumi T, Hashimoto N, Hayashi K, Kawanishi N, Fukasawa K, Takaki T,Ikeura C, Ikuta M, Suzuki-Takahashi I, Hayama T, Nishimura S and Morishima H J. Med. Chem 44 2001
Design of Compounds with a Cis Conformation¶
The aromatic phenyl was replaced by a pyridine, for all possible positions of the nitrogen atom (ortho, meta, para). The increased activity observed only for the 2-pyridyl isomer was interpreted as due to the increased population of the cis conformation (favored by an internal hydrogen bond).
articles
Structure-Based Generation of a New Class of Potent Cdk4 Inhibitors: New de Novo Design Strategy and Library Design Honma T, Hayashi K, Aoyama T, Hashimoto N, Machida T, Fukasawa K, Iwama T, Ikeura C, Ikuta M, Suzuki-Takahashi I, Iwasawa Y, Hayama T, Nishimura S and Morishima H J. Med. Chem 44 2001
A Novel Approach for the Development of Selective Cdk4 Inhibitors: Library Design Based on Locations of Cdk4 Specific Amino Acid Residues Honma T, Yoshizumi T, Hashimoto N, Hayashi K, Kawanishi N, Fukasawa K, Takaki T,Ikeura C, Ikuta M, Suzuki-Takahashi I, Hayama T, Nishimura S and Morishima H J. Med. Chem 44 2001
Good Exploitation of the SAR Analyses¶
The SAR analyses were further exploited and led to the discovery of very potent and selective compounds such as 15a. In summary, the major steps in this project were (1) discovery of a hit; (2) importance of the urea moiety; (3) modeling the probable bioactive conformation; (4) analogs favoring the cis conformation; (5) lead optimization.

articles
Structure-Based Generation of a New Class of Potent Cdk4 Inhibitors: New de Novo Design Strategy and Library Design Honma T, Hayashi K, Aoyama T, Hashimoto N, Machida T, Fukasawa K, Iwama T, Ikeura C, Ikuta M, Suzuki-Takahashi I, Iwasawa Y, Hayama T, Nishimura S and Morishima H J. Med. Chem 44 2001
A Novel Approach for the Development of Selective Cdk4 Inhibitors: Library Design Based on Locations of Cdk4 Specific Amino Acid Residues Honma T, Yoshizumi T, Hashimoto N, Hayashi K, Kawanishi N, Fukasawa K, Takaki T,Ikeura C, Ikuta M, Suzuki-Takahashi I, Hayama T, Nishimura S and Morishima H J. Med. Chem 44 2001
Case Study-2 : Dioxobenzothiazole Example¶
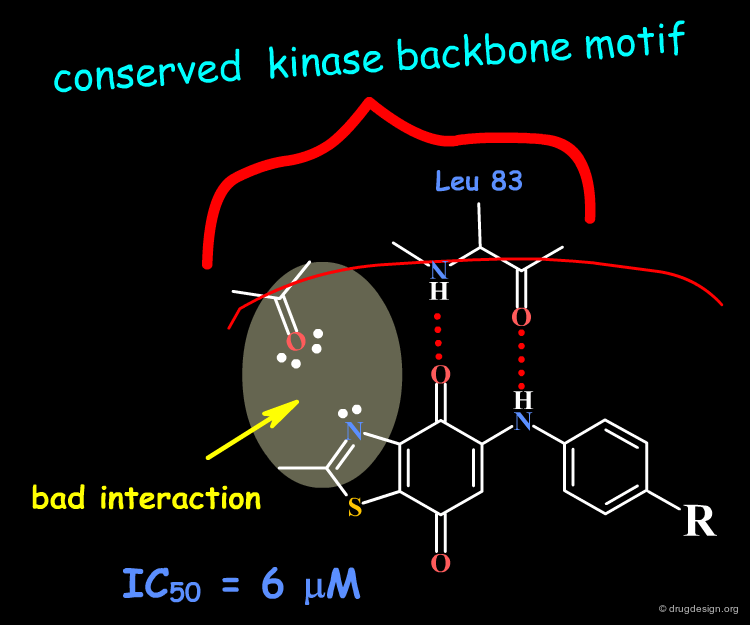
The Dioxobenzothiazole Scaffold¶
The dioxobenzothiazole scaffold has been the object of SAR studies, in a project aiming at the discovery of CDK4 kinase inhibitors. Starting with the unsubstituted derivative having an IC50 of 6 µM, a series of analogs were prepared.

articles
5-Arylamino-2-methyl-4,7-dioxobenzothiazoles as inhibitors of cyclin-dependent kinase 4 and cytotoxic agents Ryu CK, Kang HY, Lee SK, Nam KA, Hong CY, Ko WG and Lee BH Bioorg. Med. Chem. Lett 10 2000
Optimization of the Dioxobenzothiazole Lead¶
A classical optimization of the series failed to find more potent compounds.

articles
5-Arylamino-2-methyl-4,7-dioxobenzothiazoles as inhibitors of cyclin-dependent kinase 4 and cytotoxic agents Ryu CK, Kang HY, Lee SK, Nam KA, Hong CY, Ko WG and Lee BH Bioorg. Med. Chem. Lett 10 2000
SAR Analyses¶
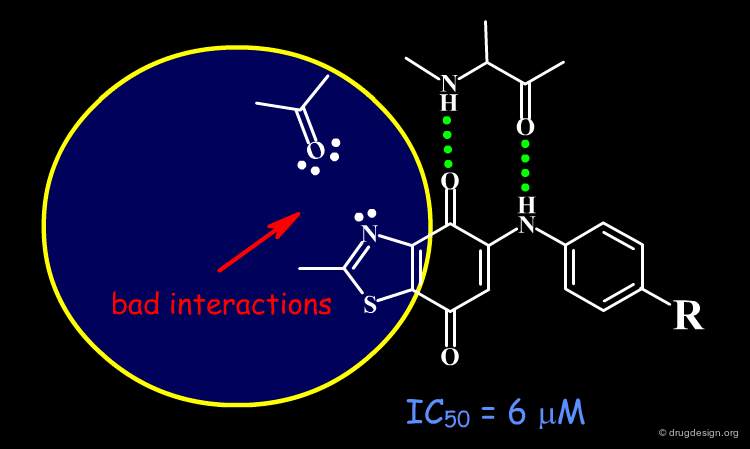
The binding of the dioxobenzothiazole series can be interpreted in the light the known bidentate hydrogen-bond binding with the conserved kinase hinge. This model provides a basis for understanding the SAR in this series and clarifies why it was not possible to find more potent analogs: the problem is due to steric and polar repulsions between the nitrogen atom of the thiazole and the carbonyl of the hinge for all the analogs prepared. Note that another scaffold containing an N-H group instead of an sp2 nitrogen atom may lead to more potent compounds.
articles
5-Arylamino-2-methyl-4,7-dioxobenzothiazoles as inhibitors of cyclin-dependent kinase 4 and cytotoxic agents Ryu CK, Kang HY, Lee SK, Nam KA, Hong CY, Ko WG and Lee BH Bioorg. Med. Chem. Lett 10 2000
Docking of the Dioxobenzothiazole Molecule¶
Docking analyses confirm that there are undesirable steric and polar repulsions between the nitrogen atom of the thiazole, the methyl group and the carbonyl of the hinge, a characteristic present in all analogs synthesized.

Being Trapped with a Bad Scaffold¶
When a lead is chosen, it is useful to make sure that the corresponding scaffold does not contain structural elements that are constantly detrimental to the biological activities. It is worth spending some time addressing this issue before embarking on an SAR optimization program with a bad scaffold.
Case Study-3 : EGF-R Kinase Inhibitors¶
Therapeutic Utility of EGF-R Kinase Inhibitors¶
Inhibitors of the EGF-R protein tyrosine kinase have enormous therapeutic potential in the treatment of malignant and nonmalignant epithelial diseases. Historically, this protein is one of the first tyrosine kinases described in the literature and was therefore chosen by many companies as a target for a research program in the signal transduction area. Many inhibitors from several structurally diverse classes were discovered, making it possible to interpret the SAR observed for these molecules.

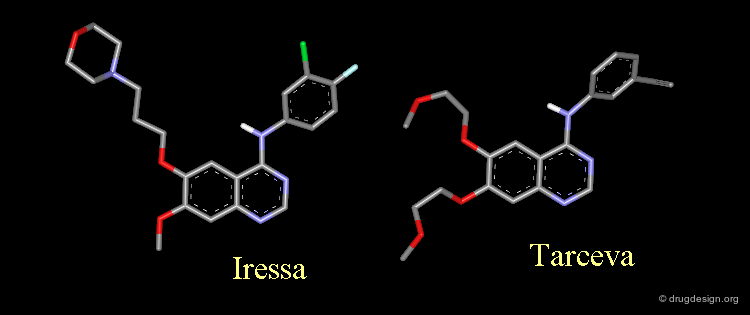
Amino-4 Quinazoline Inhibitors: Iressa and Tarceva¶
Iressa and Tarceva are examples of inhibitors of the EGF-R protein kinase. They share the same 4-amino-quinazoline scaffold.

Analysis of Tarceva Binding to the EGF-R Kinase (1/4)¶
ATP is the common cofactor of all protein kinases and is attached to the enzyme in a bidentate manner.

articles
Structure of the Epidermal Growth Factor Receptor Kinase Domain Alone and in Complex with a 4-Anilinoquinazoline Inhibitor Stamos J, Sliwkowski MX, Eigenbrot C J Biol Chem. 277 2002
Analysis of Tarceva Binding to the EGF-R Kinase (2/4)¶
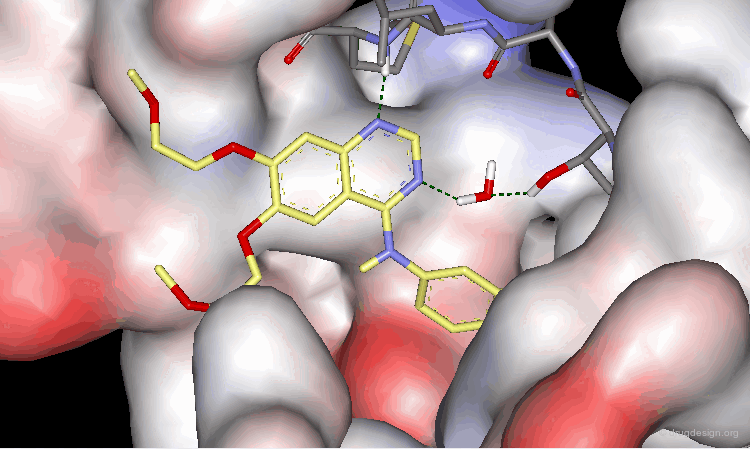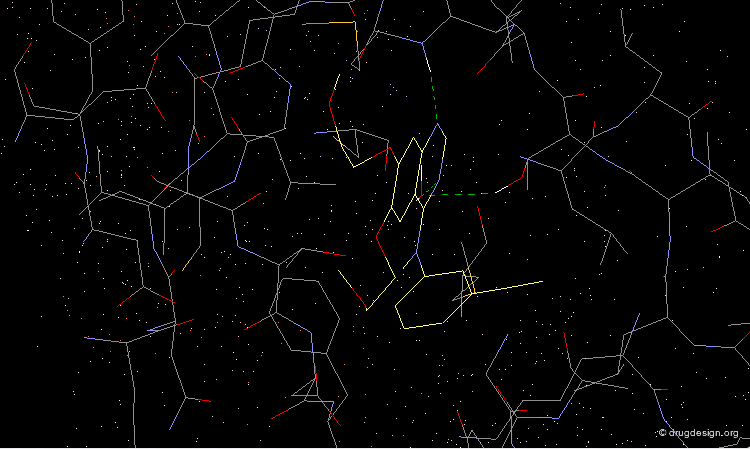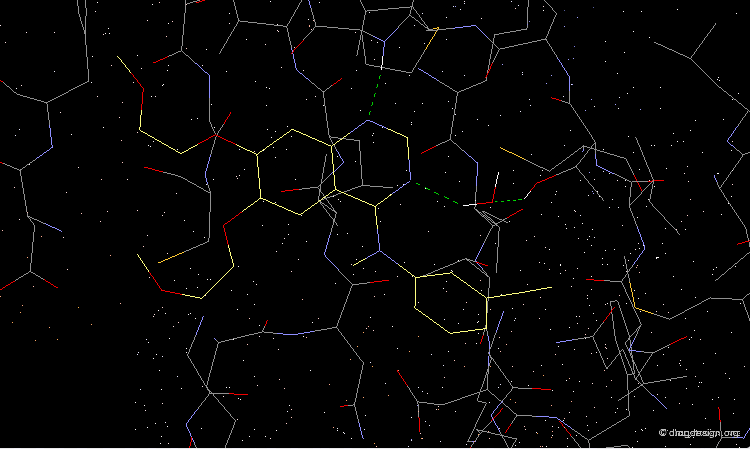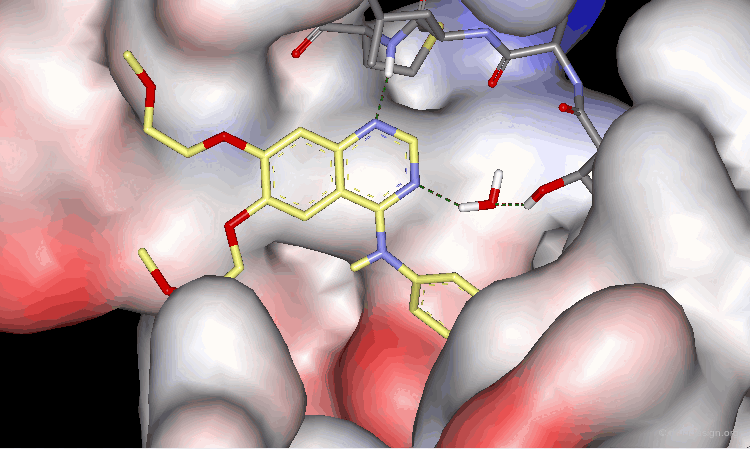
As demonstrated by X-ray studies, Tarceva adopts an extended conformation. Both nitrogen atoms of the quinazoline scaffold are involved as acceptors of hydrogen bonds; however the N3 nitrogen atom is too distant for the formation of a direct hydrogen bond with the kinase.
articles
Structure of the Epidermal Growth Factor Receptor Kinase Domain Alone and in Complex with a 4-Anilinoquinazoline Inhibitor Stamos J, Sliwkowski MX, Eigenbrot C J Biol Chem. 277 2002
Analysis of Tarceva Binding to the EGF-R Kinase (3/4)¶
Tarceva is an ATP competitive inhibitor of the EGF-R kinase. ATP and Tarceva exploit different anchoring interactions; a water molecule makes possible the interaction between the inhibitor and the enzyme.

articles
Structure of the Epidermal Growth Factor Receptor Kinase Domain Alone and in Complex with a 4-Anilinoquinazoline Inhibitor Stamos J, Sliwkowski MX, Eigenbrot C J Biol Chem. 277 2002
Analysis of Tarceva Binding to the EGF-R Kinase (4/4)¶
The substituents in positions 6 and 7 extend towards the outside of the protein and serve to optimize the physico-chemical properties for better solubility and bioavailability.

articles
Structure of the Epidermal Growth Factor Receptor Kinase Domain Alone and in Complex with a 4-Anilinoquinazoline Inhibitor Stamos J, Sliwkowski MX, Eigenbrot C J Biol Chem. 277 2002
SAR of the Quinazoline Scaffold¶
Many chemical modifications of the amino-4 quinazoline scaffold have been studied and the resulting SAR features are summarized in the figure below.

articles
Tyrosine Kinase Inhibitors. 5. Synthesis and structure-activity relationships for 4-[(phenylmethyl)amino]- and 4-(phenylamino)quinazolines as potent adenosine 5'-triphosphate binding site inhibitors of the tyrosine kinase domain of the epidermal growth factor receptor. Rewcastle GW, Denny WA, Bridges AJ, Zhou H, Cody DR, McMichael A, Fry DW J. Med. Chem. 38(18) 1995
Tyrosine Kinase Inhibitors. 8. An unusually steep structure-activity relationship for analogues of 4-(3-bromoanilino)-6,7-dimethoxyquinazoline (PD 153035), a potent inhibitor of the epidermal growth factor receptor. Bridges AJ, Zhou H, Cody DR, Rewcastle GW, McMichael A, Showalter HD, Fry DW, Kraker AJ, Denny WA. J. Med. Chem. 39(1) 1996
Tyrosine Kinase Inhibitors. 9. Synthesis and evaluation of fused tricyclic quinazoline analogues as ATP site inhibitors of the tyrosine kinase activity of the epidermal growth factor receptor. Rewcastle GW, Palmer BD, Bridges AJ, Showalter HD, Sun L, Nelson J, McMichael A, Kraker AJ, Fry DW, Denny WA. J. Med. Chem. 39(4) 1996
Tyrosine Kinase Inhibitors. 16. 6,5,6-tricyclic benzothieno inverted question mark3, 2-dpyrimidines and pyrimido inverted question mark5,4-b- and - inverted question mark4,5-bindoles as potent inhibitors of the epidermal growth factor receptor tyrosine kinase. Showalter HD, Bridges AJ, Zhou H, Sercel AD, McMichael A, Fry DW. J. Med. Chem. 42(26) 1999
The synthesis and SAR of new 4-(N-alkyl-N-phenyl)amino-6,7-dimethoxyquinazolines and 4-(N-alkyl-N-phenyl)amino-pyrazolo[3,4-d]pyrimidines, inhibitors of CSF-1R Tyrosine kinase activity Myers MR, Setzer NN, Spada AP, Persons PE, Ly CQ, Maguier MP, Zulli AL, Cheney DL, Zilberstein A, Johnson SE, Franks CF and Mitchell KJ Bioorg. Med. Chem. Lett. 7(4) 1997
Chemical Inhibitors of Protein kinases. Bridges AJ Chem. Rev. 101 2001
4-Anilino-6,7-dialkoxyquinoline-3-carbonitrile Inhibitors of Epidermal Growth Factor Receptor Kinase and Their Bioisosteric Relationship to the 4-Anilino-6,7-dialkoxyquinazoline Inhibitors. Wissner A, Berger DM, Boschelli DH, Floyd MB Jr., Greenberger LM, Gruber BC, Johnson BD, Mamuya N, Nilakantan R, Reich MF, Shen R, Tsou HR, Upeslacis E, Wang YF, Wu B, Ye F, Zhang N. J. Med. Chem. 43 2000
SAR of Fused Rings in the Quinazoline Scaffold¶
The SAR features of fused-ring additions are summarized in this diagram as well.

Analysis of a Surprising Observation¶
The AstraZeneca group replaced the phenyl of the anilino moiety by a fused bicyclic heterocycle having a nitrogen atom located in different places. They observed that a nitrogen atom para or ortho to the N4 is detrimental to potency (compounds 15 and 16).
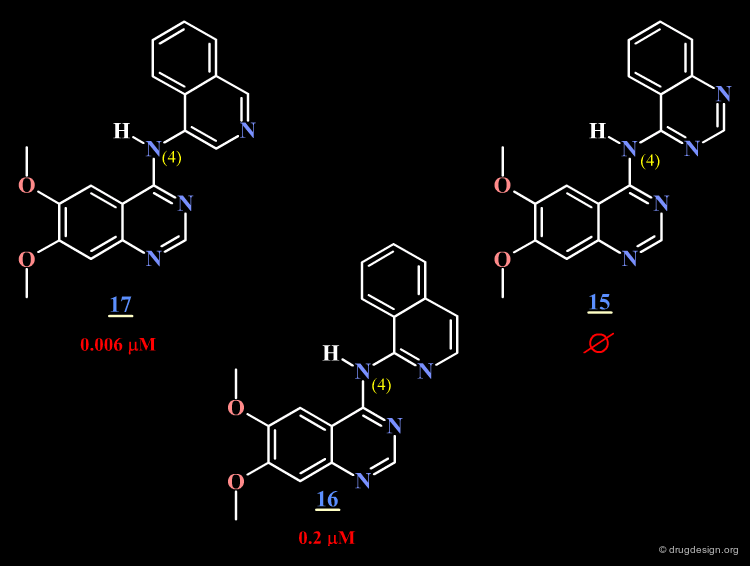
articles
Epidermal Growth Factor Receptor Tyrosine Kinase: Structure-Activity Relationships and Antitumour Activity of Novel Quinazolines Gibson KH, Grundy W, Godfery AA, Woodburn JR, Ashton SE, Curry BJ, Scarlett L, Barker AJ and Brown DS Bioorganic and Medicinal Chemistry Letters 7 1997
Analysis of Atomic Charges in the Different Analogs¶
The surprising difference in the biological activities observed for 15, 16 and 17 can be understood with 3D analyses. The rationale is only summarized in 2D to make it easier to understand the 3D view presented on the next page. For compounds 15 and 16, and due to the influence of nitrogen atoms on the electronic distribution, a positive charge is induced on the atom meta to N4, whereas in molecule 17 this position is negatively charged.
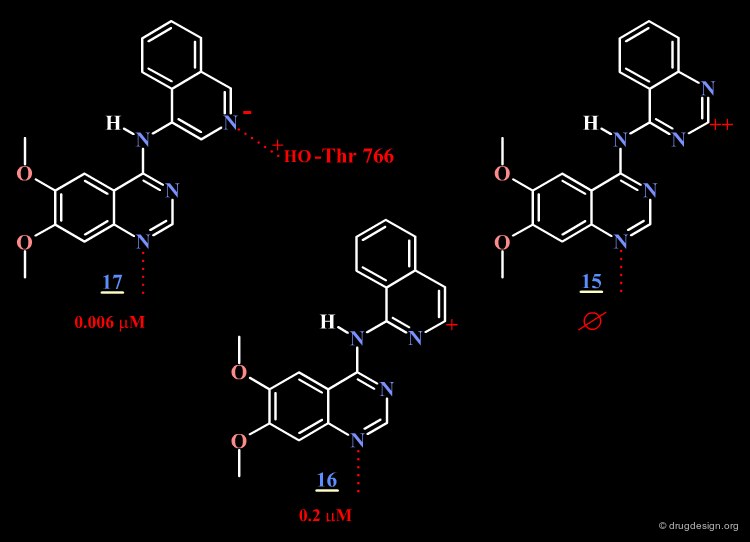
Optimal Binding of Inhibitor 17¶
The good location of the isoquinoline nitrogen atom for direct formation of a hydrogen bond and a very favorable geometry allow for optimal binding of the potent inhibitor 17 to the EGF-R kinase. A water molecule is not needed any more (as for Tarceva) to assist to the interaction of the inhibitor with the enzyme.

Case Study-4 : Nifedipine Example¶
Two Inactive Analogs of Nifedipine¶
Nifedipine is a well-known calcium channel antagonist drug used in the treatment of cardiovascular disorders including hypertension. When a methyl group is added to the structure of nifedipine either in the para position of the free phenyl ring (analog 1), or in position 4 of the 1,4-dihydropyridine ring (analog 2), the corresponding molecules are biologically inactive. Analyses of the origin of these inactivities will enable us to understand the molecular determinants that are crucial to the biological activity of nifedipine analogs.

articles
1,4-dihydropyridine calcium channel ligands: Selectivity of action. The roles of pharmacokinetics, state-dependent interactions, channel isoforms, and other factors David J. Triggle Drug Development Research 58 (1) 2003
Analysis of the 4' Substituted Analogs of Nifedipine¶
When a substituent is introduced at the 4' position of the nifedipine structure, the corresponding molecule is biologically inactive. This is thought to be caused by steric clashes with the receptor.
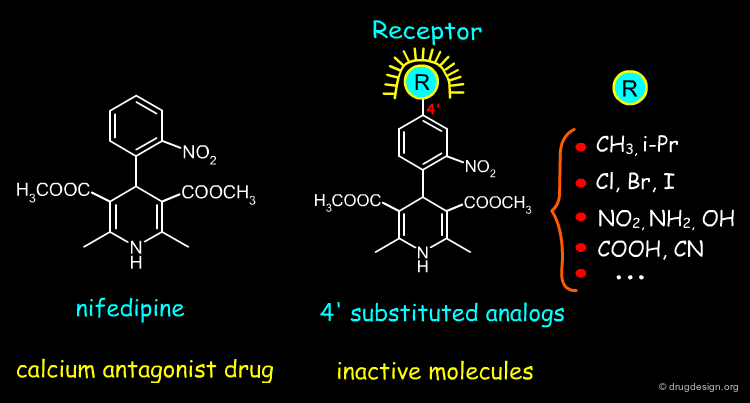
articles
1,4-dihydropyridine calcium channel ligands: Selectivity of action. The roles of pharmacokinetics, state-dependent interactions, channel isoforms, and other factors David J. Triggle Drug Development Research 58 (1) 2003
Analysis of the 4 Substituted Analogs of Nifedipine¶
The inactivity of methyl-4 nifedipine cannot reasonably be explained by the presence of steric clashes with the receptor. In this case more subtle changes occur in the structure of the molecule. To analyze them, we will first look at the molecular geometry of the phenyl-4 dihydropyridine structure.

Molecular Geometry of Phenyl-4 Dihydropyridine¶
In phenyl-4 dihydropyridine, the 1,4-dihydropyridine ring can exist in two alternative boat conformations: the phenyl substituent is either in the axial or in the equatorial orientation. In the absence of substituents at the 3 and 5 positions of that ring, the phenyl substituent prefers to be in the equatorial position, as generally found in phenyl substituted rings.

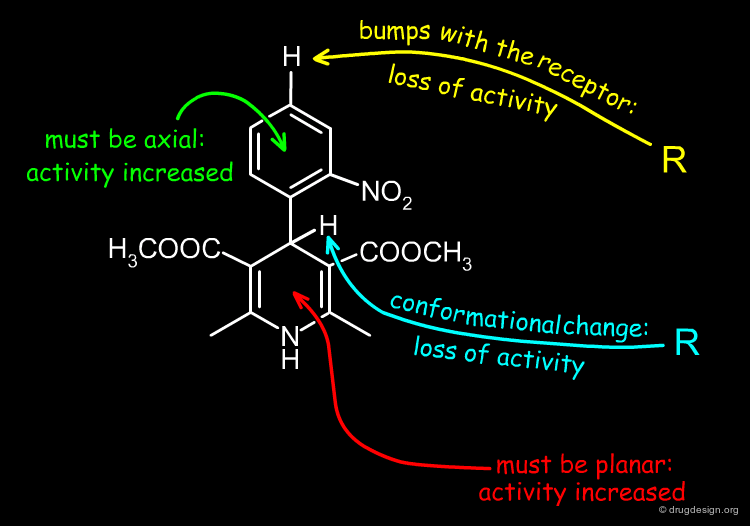
Preferred Conformation of Nifedipine¶
Contrary to the rule stated on the previous page, the preferred conformation of nifedipine corresponds to the one with the nitrophenyl in the axial position because this orientation enables the molecule to avoid the steric clashes that are created between the nitrophenyl group and the methylester substituents at the 3 and 5 positions.

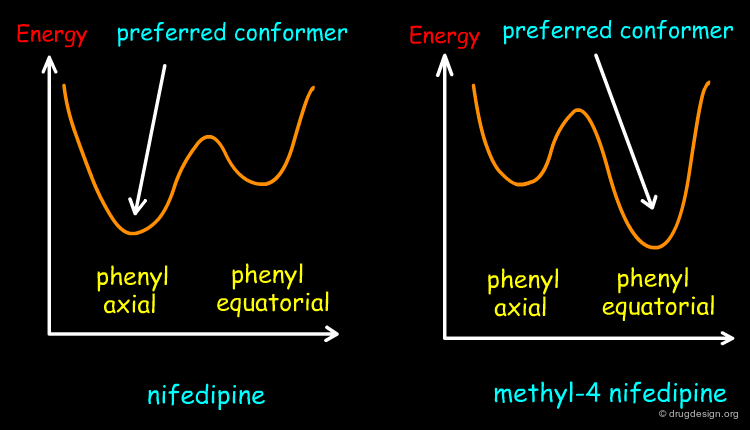
Preferred Conformation of Methyl-4 Nifedipine¶
The presence of the methyl substituent at position 4 of the dihydropyridine ring changes the conformational preference of the methyl-4 nifedipine molecule, and in this case, the preferred conformation of the molecule corresponds to the equatorial orientation of the nitrophenyl group. This has been confirmed by theoretical calculations and by X-ray crystallography. The energetic profile of methyl-4 nifedipine is compared to nifedipine in the figure below.

Bioactive Conformation of Nifedipine-Like Antagonists¶
It has been suggested that the bioactive conformation of dihydropyridine calcium antagonists corresponds to the molecular geometry with the phenyl substituent in the axial orientation. Further on the basis of detailed X-ray analyses it has been shown that the biological potencies can be correlated with the magnitude of the 1,4-dihydropyridine ring puckering: the more active compounds exhibit the smallest degree of ring distortion from planarity.

articles
Crystal structures and pharmacological activity of calcium channel antagonists: 2,6-dimethyl-3,5-dicarbomethoxy-4-(unsubstituted, 2-methyl-, 4-methyl-, 3-nitro-, 4-nitro-, and 2,4-dinitrophenyl)-1,4-dihydropyridine Fossheim R, Svarteng K, Mostad A, Romming C, Shefter E, Triggle DJ. J.Med.Chem 25 (2) 1982
Molecular-Level Model For The Agonist Antagonist Selectivity Of The 1,4-Dihydropyridine Calcium-Channel Receptor Langs, D.A., Kwon, Y.W., Strong, P.D., Triggle, D.J Journal of Computer-Aided Molecular Design 5 1991
1,4-Dihydropyridines as Calcium Channel Ligands and Privileged Structures Triggle DJ Cellular and Molecular Neurobiology 23 (3) 2003
SAR Analyses Require Great Attention¶
The examples presented in this chapter clearly demonstrate that structure-activity relationship data need to be analyzed in minute detail and carefully differentiated. Then, general rules can be deduced to get a good grasp of the structural determinants governing the biological properties of the molecules.
articles
Crystal structures and pharmacological activity of calcium channel antagonists: 2,6-dimethyl-3,5-dicarbomethoxy-4-(unsubstituted, 2-methyl-, 4-methyl-, 3-nitro-, 4-nitro-, and 2,4-dinitrophenyl)-1,4-dihydropyridine Fossheim R, Svarteng K, Mostad A, Romming C, Shefter E, Triggle DJ. J.Med.Chem 25 (2) 1982
Molecular-Level Model For The Agonist Antagonist Selectivity Of The 1,4-Dihydropyridine Calcium-Channel Receptor Langs, D.A., Kwon, Y.W., Strong, P.D., Triggle, D.J Journal of Computer-Aided Molecular Design 5 1991
1,4-Dihydropyridines as Calcium Channel Ligands and Privileged Structures Triggle DJ Cellular and Molecular Neurobiology 23 (3) 2003
Case Study-5 : Carbonic Anhydrase Example¶
The Carbonic Anhydrase Protein¶
Carbonic anhydrase catalyzes the reversible reaction involving hydration of carbon dioxide and dehydration of carbonic acid. There are a number of isozymes of carbonic anhydrase. The isozyme, CA-II is found in red blood cells and other cells including the ciliary body of the eye. All the isoenzymes have a molecular weight of approximately 30,000 and contain one zinc atom per molecule. In the following pages we present how structure-activity relationship analyses led to the discovery of inhibitors of Carbonic anhydrase with sub-nanomolar activities.

Therapeutic Utility of CA Inhibitors¶
Carbonic anhydrase inhibitors are involved in multiple actions in the body. They decrease the formation of aqueous humor (the fluid found in the anterior chamber of the eye), resulting in decreased intraocular pressure, helping in the treatment of glaucoma. They also cause a diuretic type of action in the kidneys, which increases the amount of urine excreted. Carbonic anhydrase inhibitors appear to have some effect on the central nervous system (CNS) and have been found to be useful in the treatment of certain CNS disorders, such as epilepsy, when used together with other appropriate medications.

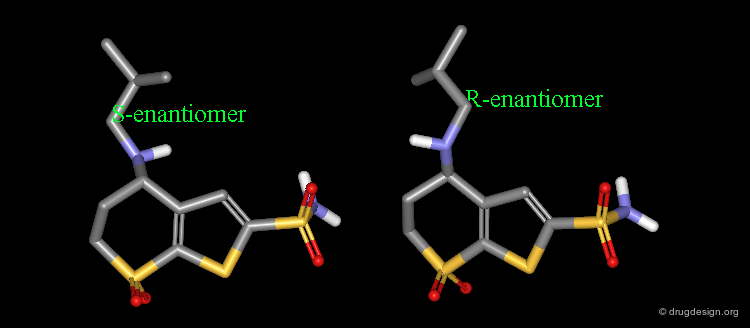
MK-417 is a Potent Inhibitor of CA¶
MK-417 is a potent carbonic anhydrase inhibitor (pKi = 0.61 nM), which rapidly penetrates into the ocular tissues after topical application. The R-enantiomer of MK-417 is about 100 times less potent (pKi = 71 nM).

articles
Thienothiopyran-2-Sulfonamides; Novel Topically Active Carbonic Anhydrase Inhibitors for the Treatment of Glaucoma Baldwin JJ, Ponticello GS, Anderson PS, Christy ME, Murcko MA., Randall WC, Schwam H, Sugrue M.F, Springer JP, Grove J., Mallorga P, Viader M-P, McKeever BM and Navia MA J. Med. Chem. 32 1989
Binding of MK-417 with CA Protein (1/5)¶
The X-ray structures of the complexes show that the binding mode of the two enantiomers is similar: The sulfonamide group is rigidly locked in a very small pocket at the back of the site interacting with a zinc atom which is tetrahedrally coordinated by three histidines.
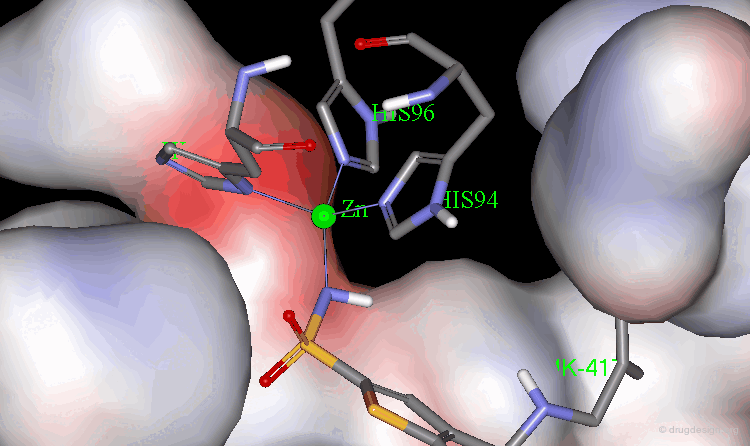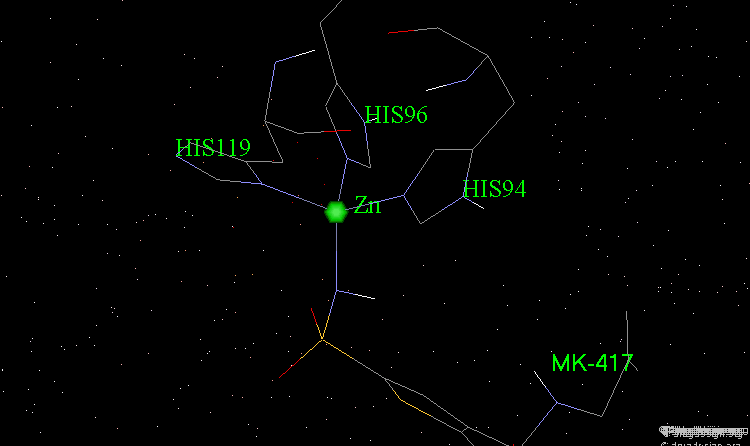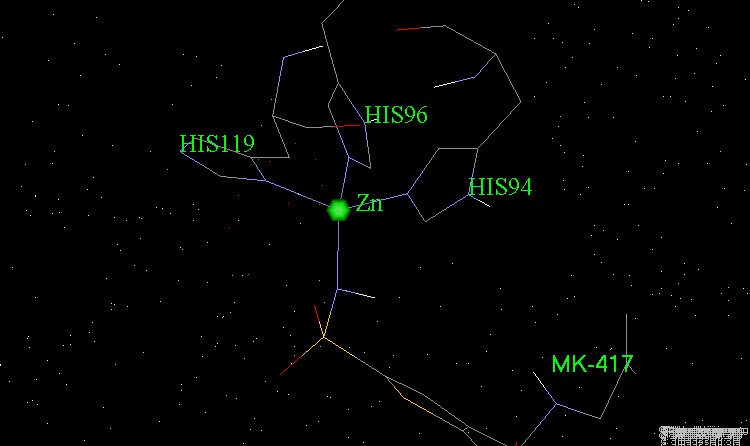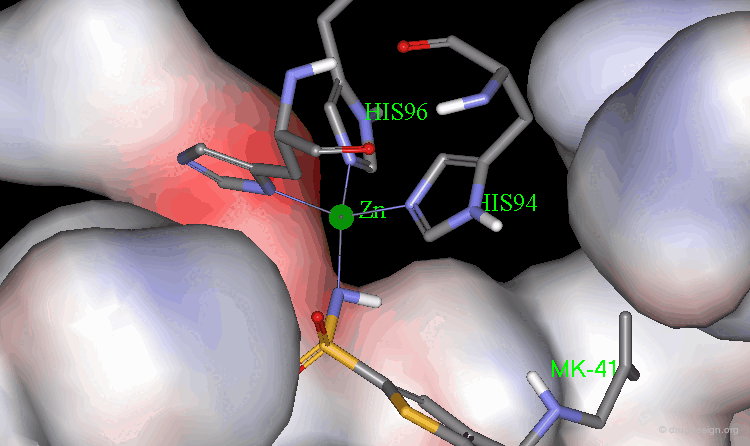

Binding of MK-417 with CA Protein (2/5)¶
The ring system and side chain lie across a larger cavity, but the positions of the sulfone group and the ethyl group are strongly determined by the way they fit precisely into small niches in the cavity.

Binding of MK-417 with CA Protein (3/5)¶
Both enantiomers placed the alkyl amino group in the less favored pseudo-axial orientation.

Binding of MK-417 with CA Protein (4/5)¶
Hydrogen bonds are formed between both enantiomers and the enzyme involving the oxygen of the SO2 group and the amino acid Gln-92.

Binding of MK-417 with CA Protein (5/5)¶
Hydrogen bonds are formed between both enantiomers and the enzyme involving the amino acids Thr-199 and Thr-200.

S Enantiomer is more Potent than the R¶
The X-ray of both complex MK-417 and its enantiomer show a good superimposition between the two ligands. Why is MK-417 100 times more potent than its enantiomer ?

Geometry of Isobutyl Side Chain by X-rays¶
The X-ray structures suggest that in the bioactive conformation of MK-417 the isobutyl side chain has an anti geometry whereas in the case of its enantiomer the isobutyl side chain has a gauche geometry.
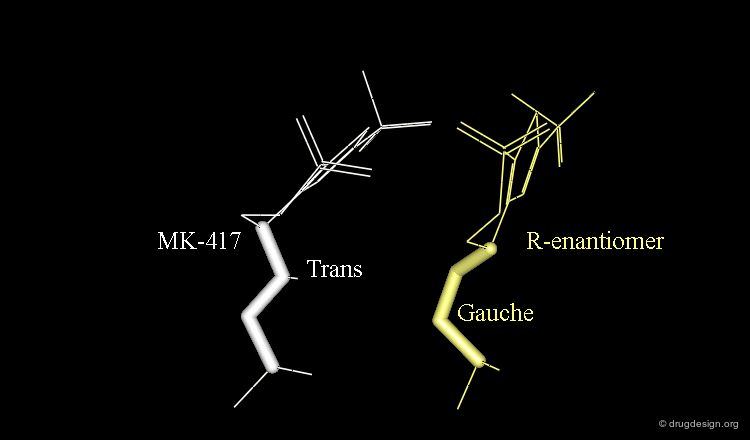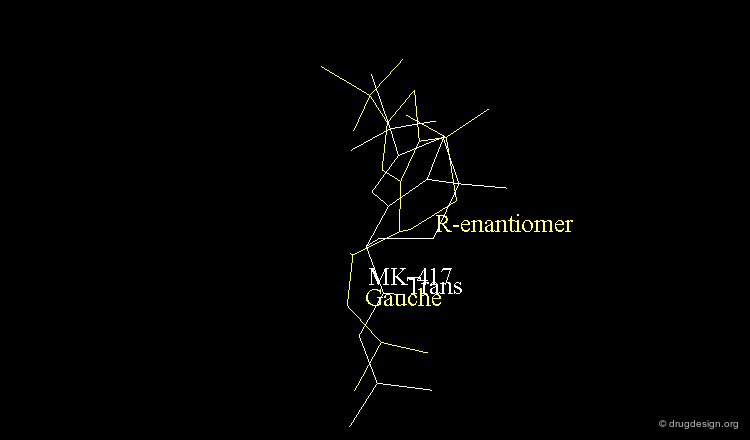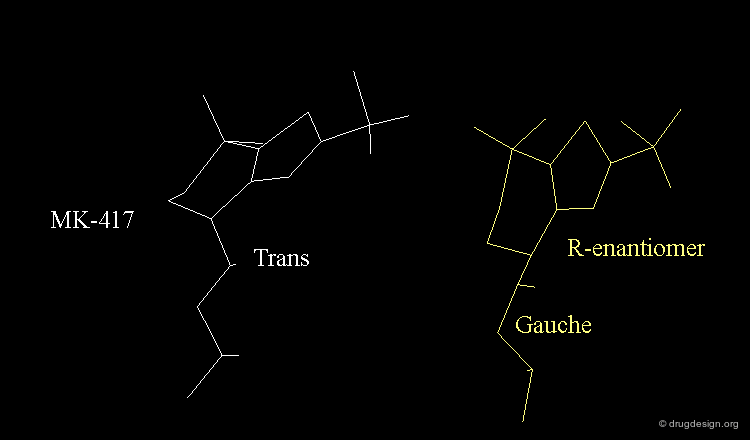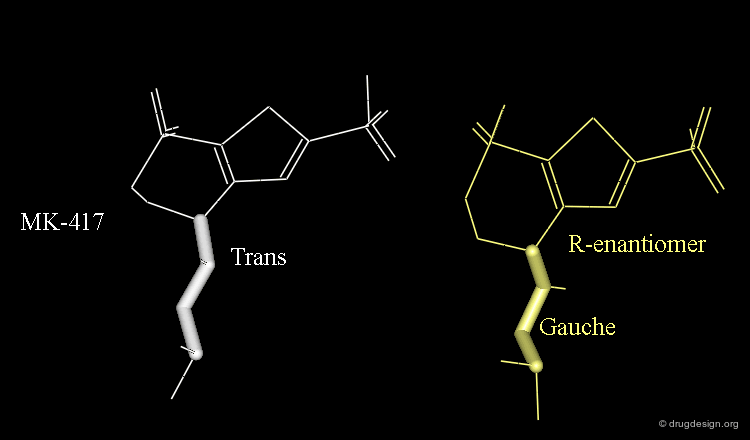
Axial/Equatorial Orientation of the Side Chain¶
In MK-417 the side chain is axial but equatorial for the R-enantiomer.
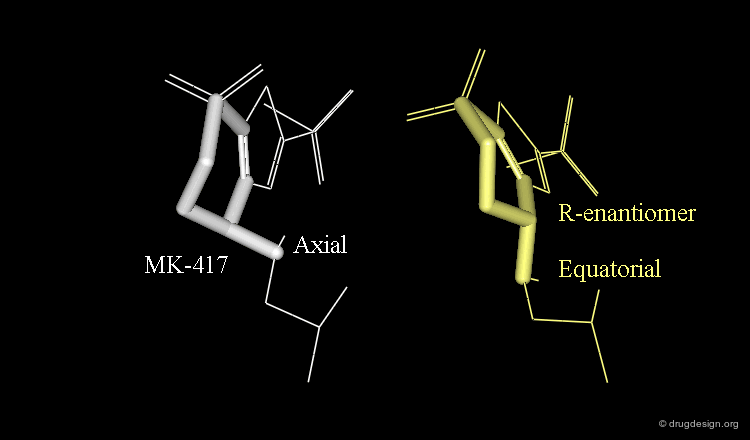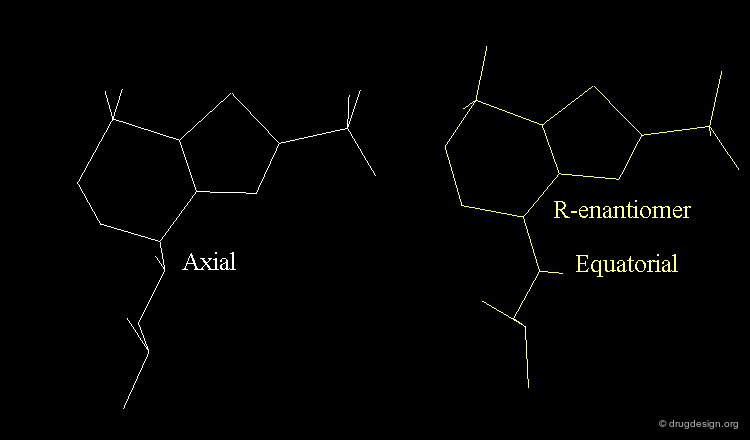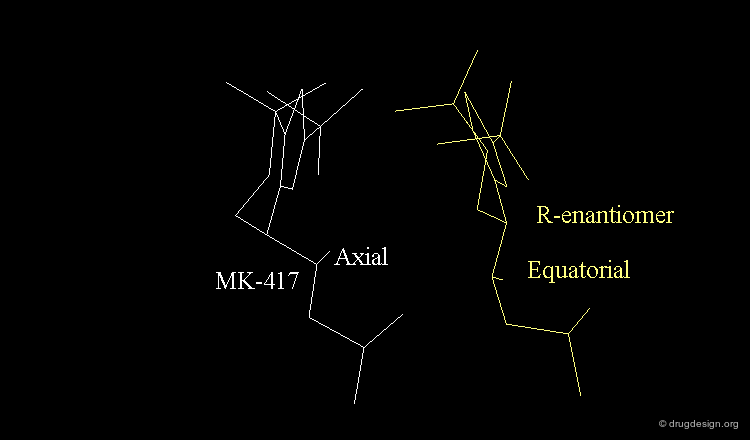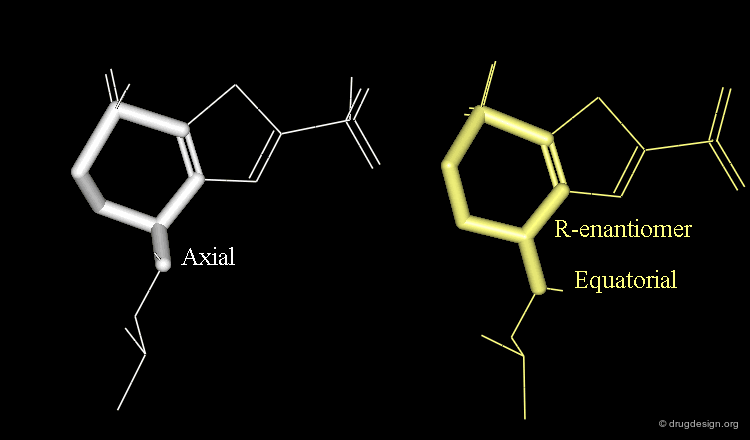
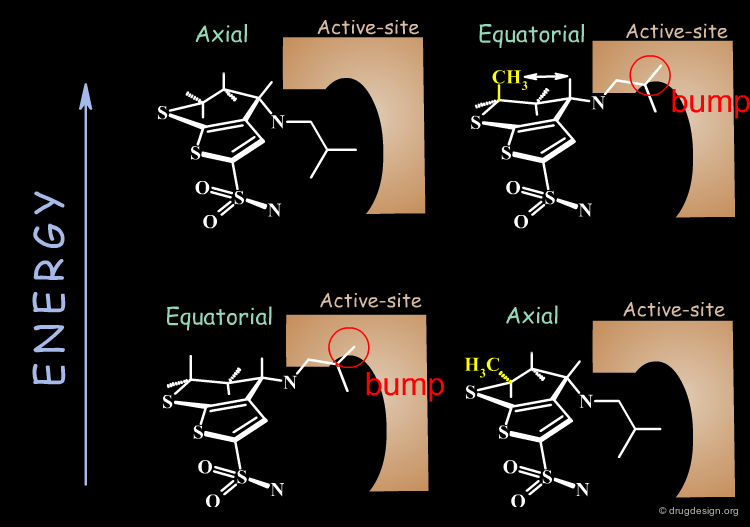
Bioactive Conformation of MK-417¶
Why is the bioactive conformation of MK-417 axial ? MK-417 can adopt two conformations with axial and equatorial side chain. The equatorial conformation energy is 4.2 kJ/mol lower than the axial conformation. But equatorial conformation does not fit in the active site.

Energy Considerations¶
Why does the R-enantiomer have an equatorial-gauche conformation? The two possible conformations, axial and the lower energy equatorial of R-enantiomer, cannot fit into the active site. To enter into the active site, the equatorial conformation must adopt a less energetically favored gauche conformation.
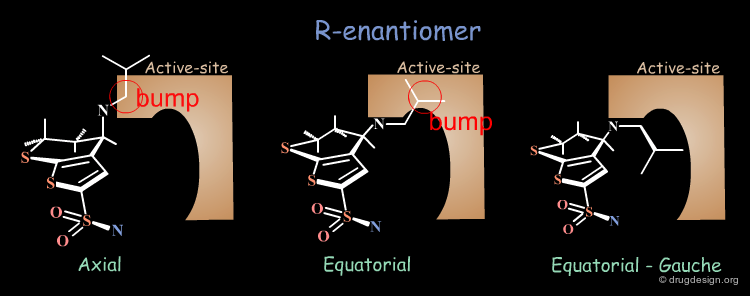
Results of the Analyses¶
Conclusion: calculations show that the MK-417(S-axial-trans) is lower in energy than its enantiomer R-equatorial-gauche. This explains why MK-417 is more potent than its R-enantiomer.
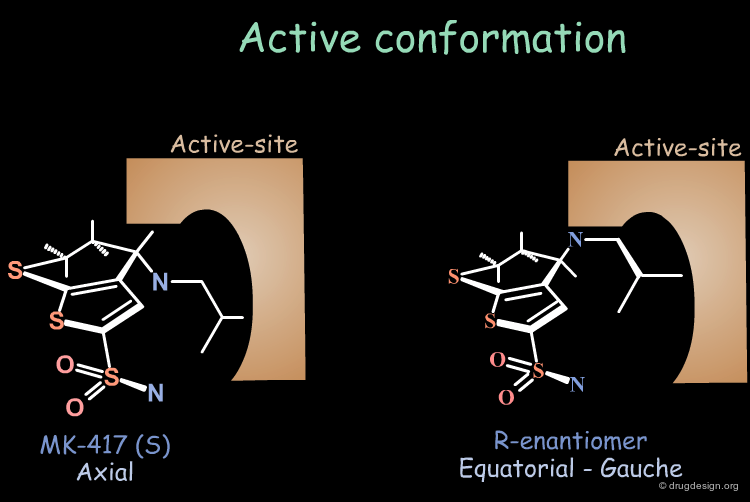
Optimization of MK-417¶
In the bioactive conformation of MK-417, the isobutylamino group is oriented in the less favored pseudo-axial conformation. By adding a methyl group in position 6 (pro-S), this conformation becomes energetically favored.
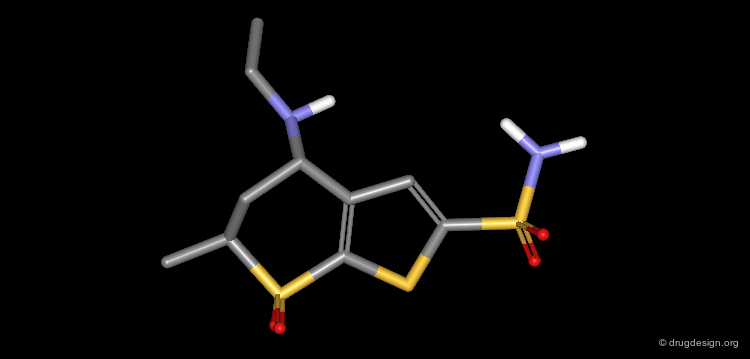
Dorzolamide, a Potent Inhibitor of CA¶
These modifications lead to a new thienothiopyran analog (dorzolamide). To preserve the physicochemical properties of the molecule, two methyl substituents were removed from the isobutyl side chain. The potency was slightly improved (pKi = 0.37 nM).


Complex of CA with Dorzolamide¶
The crystal structure of the complex CA/Dorzolamide has been resolved and is visualized here with some details.




articles
Positions of His-64 and a Bound Water in Human Carbonic Anhydrase II upon Binding Three Structurally Related Inhibitors Smith GM, Alexander RS, Christianson DW, McKeever BM, Ponticello GS, Springer JP, Randall WC, Baldwin JJ and Habecker CN Protein Sci. 3 1994
Binding of Dorzolamide with CA Protein (1/5)¶
The X-ray structure indicates that the bioactive conformation of dorzolamide fulfills the geometric requirements needed to be a potent carbonic anhydrase inhibitor. The sulfonamide group interacts with the zinc atom.
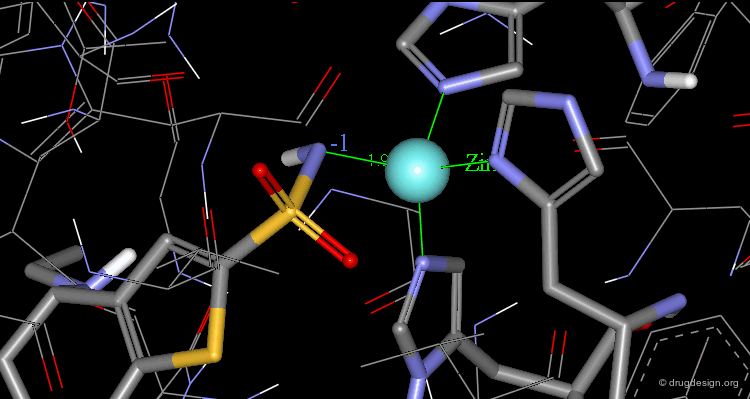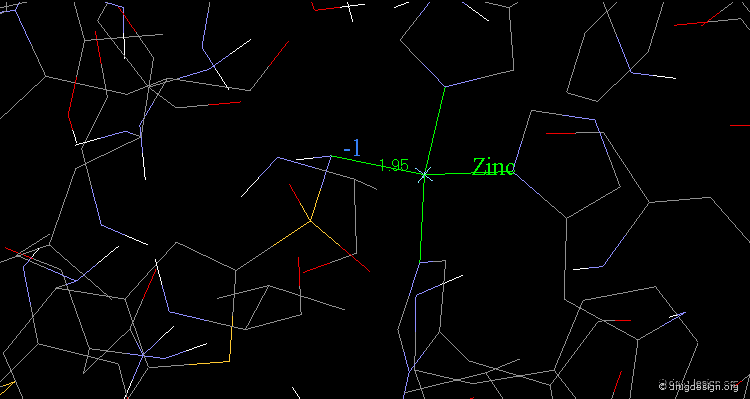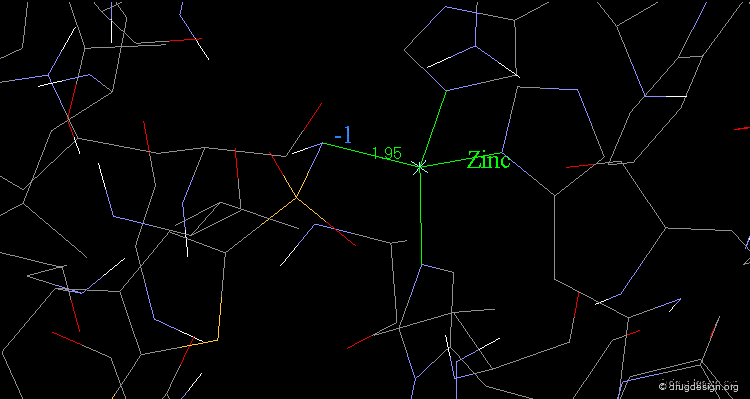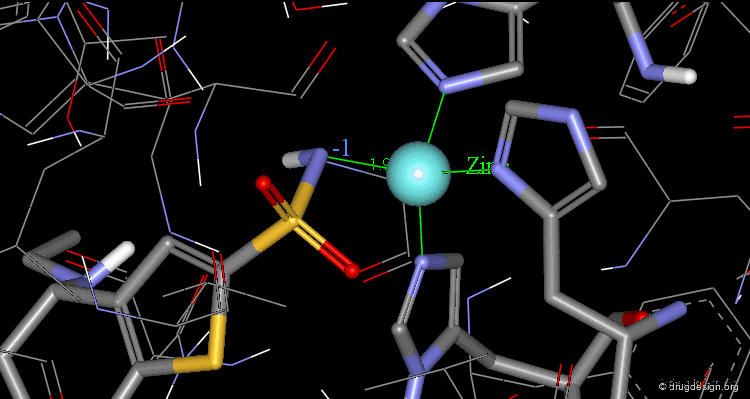
articles
Positions of His-64 and a Bound Water in Human Carbonic Anhydrase II upon Binding Three Structurally Related Inhibitors Smith GM, Alexander RS, Christianson DW, McKeever BM, Ponticello GS, Springer JP, Randall WC, Baldwin JJ and Habecker CN Protein Sci. 3 1994
Binding of Dorzolamide with CA Protein (2/5)¶
The SO2 groups form hydrogen bonds with the enzyme

articles
Positions of His-64 and a Bound Water in Human Carbonic Anhydrase II upon Binding Three Structurally Related Inhibitors Smith GM, Alexander RS, Christianson DW, McKeever BM, Ponticello GS, Springer JP, Randall WC, Baldwin JJ and Habecker CN Protein Sci. 3 1994
Binding of Dorzolamide with CA Protein (3/5)¶
The amino group in the bioactive conformation of dorzolamide is in a pseudo-axial orientation.

articles
Positions of His-64 and a Bound Water in Human Carbonic Anhydrase II upon Binding Three Structurally Related Inhibitors Smith GM, Alexander RS, Christianson DW, McKeever BM, Ponticello GS, Springer JP, Randall WC, Baldwin JJ and Habecker CN Protein Sci. 3 1994
Binding of Dorzolamide with CA Protein (4/5)¶
The isobutyl side chain has an anti geometry.
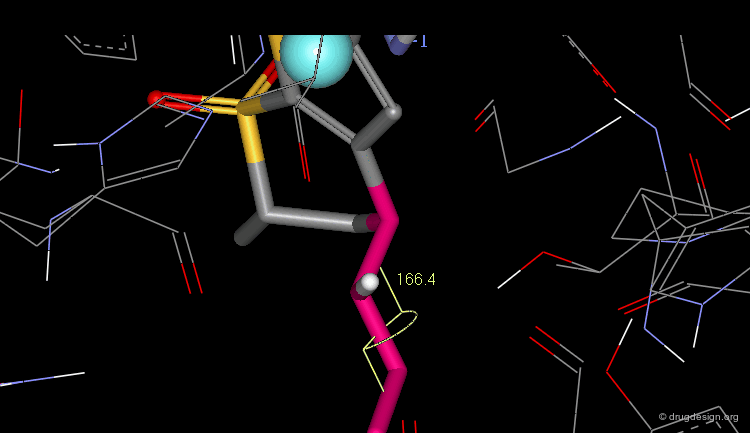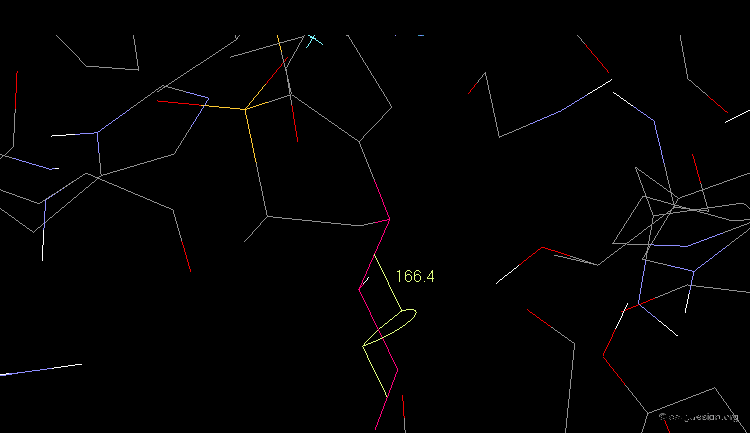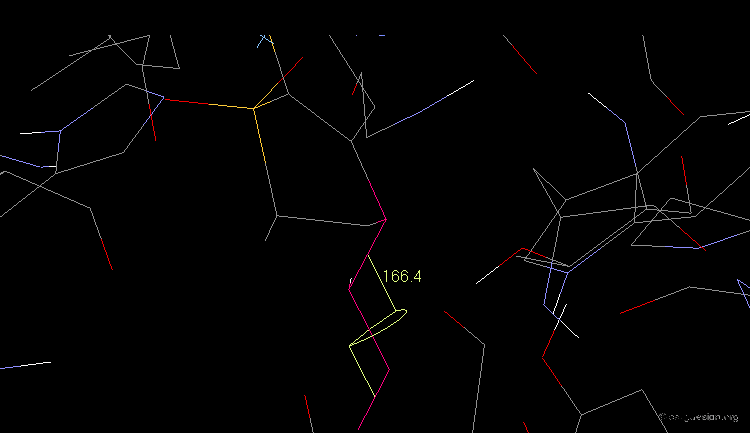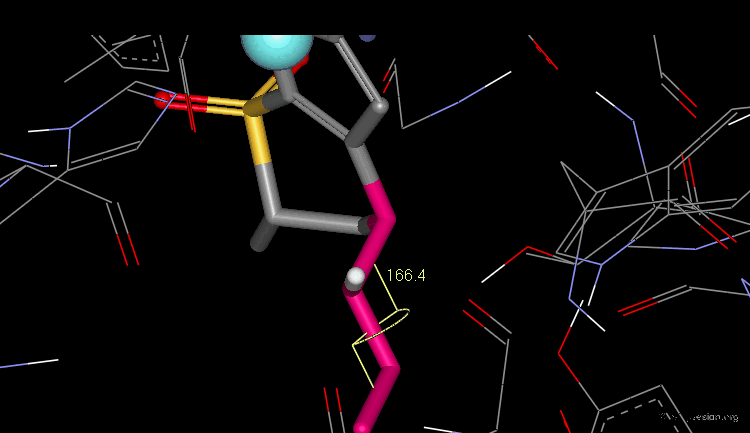
articles
Positions of His-64 and a Bound Water in Human Carbonic Anhydrase II upon Binding Three Structurally Related Inhibitors Smith GM, Alexander RS, Christianson DW, McKeever BM, Ponticello GS, Springer JP, Randall WC, Baldwin JJ and Habecker CN Protein Sci. 3 1994
Binding of Dorzolamide with CA Protein (5/5)¶
The dihedral angle between the sulfonamide group and the thiophene ring (N-S-C-S) is about 140°, which is the optimal angle.

articles
Positions of His-64 and a Bound Water in Human Carbonic Anhydrase II upon Binding Three Structurally Related Inhibitors Smith GM, Alexander RS, Christianson DW, McKeever BM, Ponticello GS, Springer JP, Randall WC, Baldwin JJ and Habecker CN Protein Sci. 3 1994
Case Study-6 : β-Lactam Antibiotics¶
Antibiotic Activities in the Cephalosporin Series¶
In the course of synthetic work on cephalosporins, analogous compounds were prepared in which the double bond was in the Δ2 position. It was observed that these cephalosporins were devoid of any antibiotic action. We describe in the following pages attempts that were made to understand the observed SAR.

articles
Beta-Lactam Antibiotics: Geometrical Requirements for Antibacterial Activities Cohen NC J.Med.Chem. 28 1983
Stereochemical Hypothesis¶
At the time this observation was made the α stereochemistry of the penicillin carboxylic function was already known from X-ray crystallography, but in the Δ2 cephalosporin this stereochemistry had not yet been proven. It was therefore suspected that the biological inactivities of the Δ2 cephalosporins were possibly due to a "wrong" configuration of this carboxylic function in the β orientation.

articles
Chemistry of Cephalosporins. XII. Configuration of the Carboxyl Group in δ-2 Cephalosporins Van Heyningen E and Ahern LK J. Med. Chem. 11 1968
Same Stereochemistry in the two Structures¶
Subsequent studies showed that the absolute configuration of the carboxy group was, in fact, α and therefore identical with the one in penicillins.

Inactivity not due to a Wrong Stereochemistry¶
The biological inactivity was then attributed to a difference in the stability of the β-lactam ring and not to a "wrong" carboxy configuration.
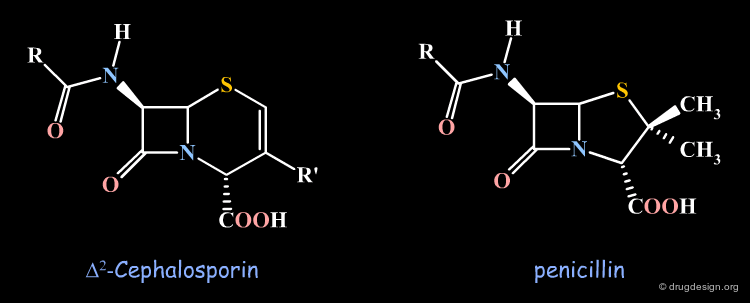
Attempts Towards a Geometrical Interpretation¶
New attempts were made by analyzing the geometrical properties and were carried out with X-ray studies. The 3D comparison of active penicillins, Δ3 and Δ2 cephalosporins was expected to reveal obvious similarities between the active compounds, penicillin and Δ3 cephalopsorin. Surprisingly it showed that penicillin more closely resembled the inactive Δ2 cephalosporin.
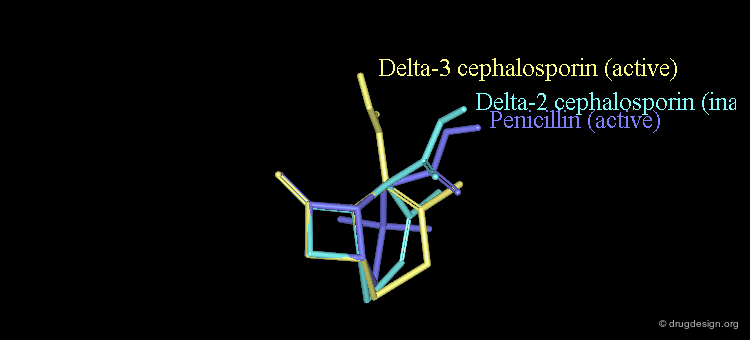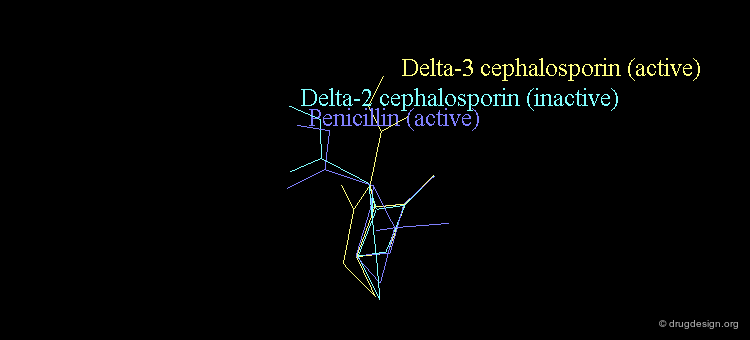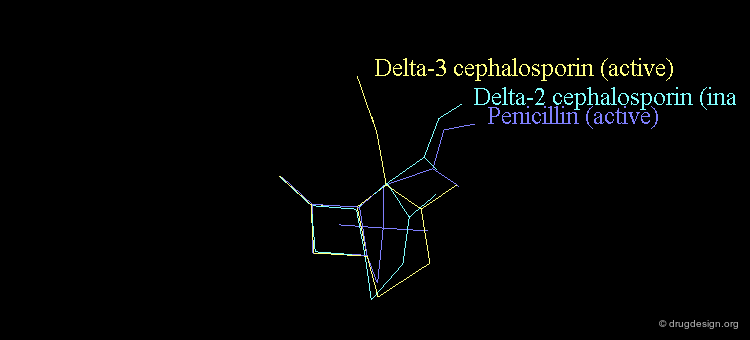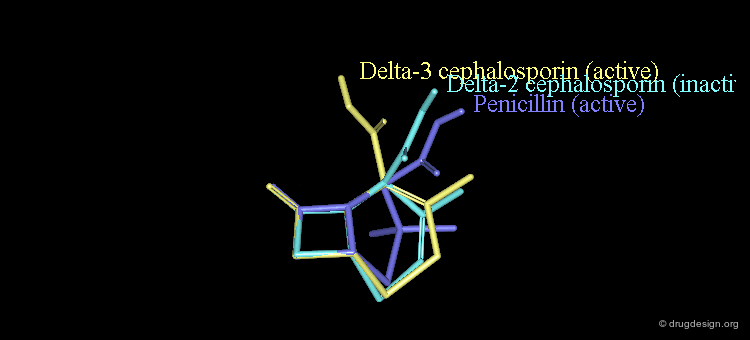
articles
A Refinement of the Crystal Structure of Potassium Benzylpenicillin Pitt GJ Acta Cryst. 5 1952
Molecular Architecture of the Cephalosporins. Insights into Biological Activity Based on Structural Investigations Sweet RM, Dahl LF J. Am. Chem. Soc. 92 1970
Flexibility of the Enzyme ?¶
It was concluded that the geometrical requirements were not very restrictive as regards identifying the antibiotics as a substrate by the enzymes. The flexibility of the enzyme was also believed to easily adapt to the geometrical features of the molecules. However, the inactivity of the Δ2 cephalosporins remained unaccounted for.

The Lactam Nitrogen Pyramidality Hypothesis¶
In the X-ray studies it was also found that the lactam nitrogen atom of the Δ2 cephalosporins was almost planar, in contrast to the pyramidal character existing in Penicillin and Δ3 cephalosporin antibiotics. The biological activities observed in the three families of compounds were again attributed to the chemical reactivity of the β-lactam amide bond.

articles
Beta-Lactam Antibiotics: Geometrical Requirements for Antibacterial Activities Cohen NC J.Med.Chem. 28 1983
Deficiency of the Nitrogen Pyramidality Hypothesis¶
While developing a thienamycin antibiotic with high antibacterial properties, Δ1-thienamycin, a double bond isomer of thienamycin, was discovered but it was devoid of any antibiotic activity. When compared to thienamycin, its inactivity was attributed to a chemically less reactive β-lactam amide bond.

articles
Structure, Reactivity, and Biological Activity of Strained Bicyclic beta.-Lactams Pfaendler HR, Gosteli J, Woodward RB, and Rihs G J. Am. Chem. Soc. 103 1981
Preparation and Antibacterial Activity of delta1-Thienamycin Shih DH and Ratcliffe RW J. Med. Chem. 24 1981
Penems and Related Substances Woodward RB Philos. Trans. R Soc. Lond. B Biol. Sci. 289 1980
Penem and Carbapenems¶
At the same time, the antibacterial activities of even simpler but similar highly strained β-lactams were studied. It was found that penem and Δ2 carbapenem possess potential antibacterial activities, whereas Δ1 carbapenem was found to be inactive.

Pyramidality of Nitrogen Atom ?¶
Analyses of the chemical reactivity of the molecules were made to assess the validity of the nitrogen pyramidality hypothesis. The ease of hydrolysis of the inactive Δ2 carbapenem compared well with that of the active parent compounds. Moreover, the pyramidality of the nitrogen atom in that molecule is highly pronounced, and this observation runs counter of the nitrogen pyramidality hypothesis.
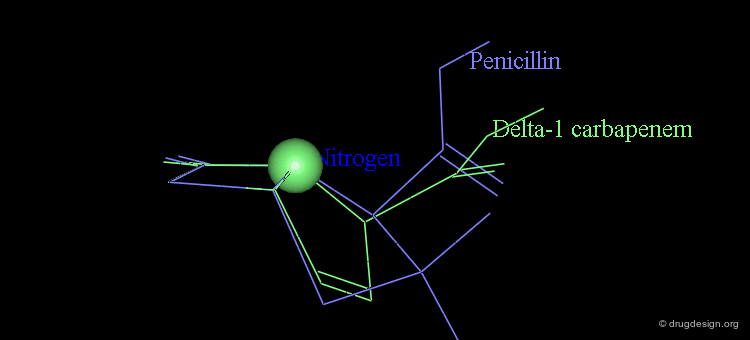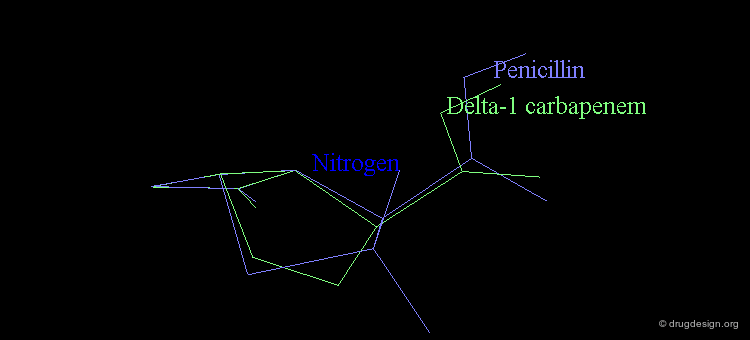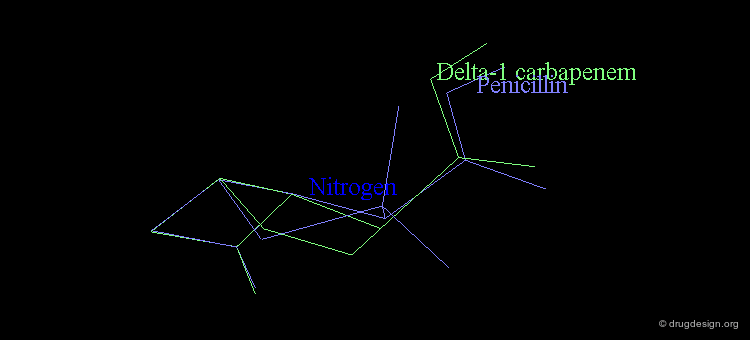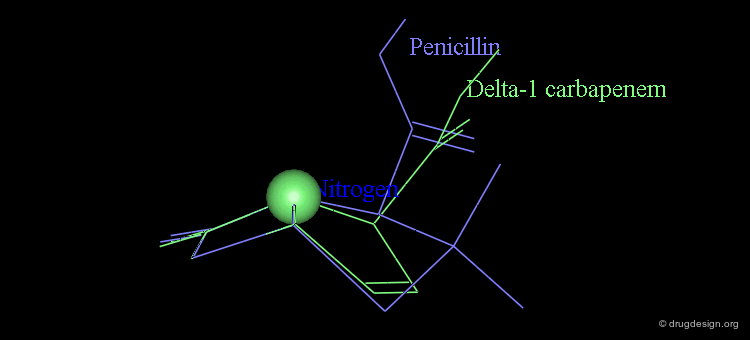
Geometrical Aspects Need to be Reassessed¶
The pyramidal character of the nitrogen atom is not sufficient to interpret the biological activities. The geometrical features, although not yet clearly understood, may play an important role in the biochemical process involved. Analyses of the geometrical features need to be reassessed.

Bioactive Conformation of Penicillins?¶
It is imperative to pay close attention to the conformational possibilities of molecules. In particular the bicyclic penam nucleus of penicillins is capable of pseudorotating movements. In this process, the cyclic system can modify the orientation of the substituents and its geometry as shown here.
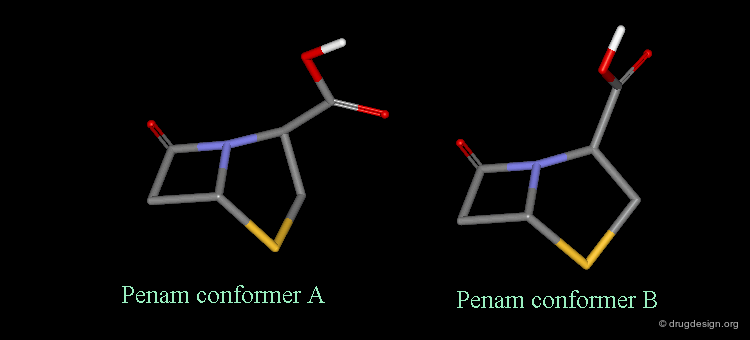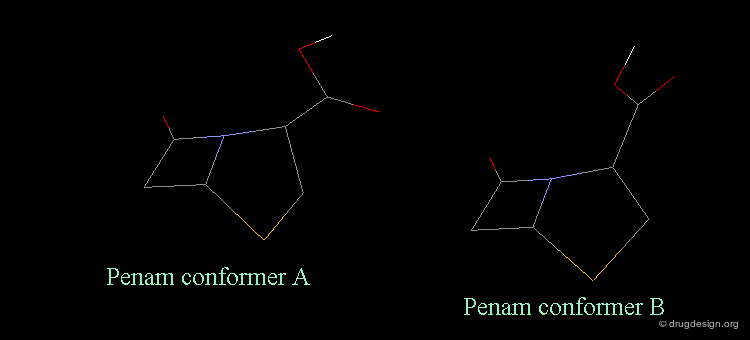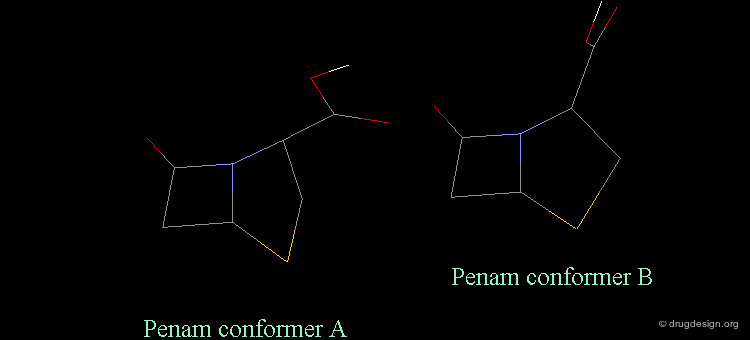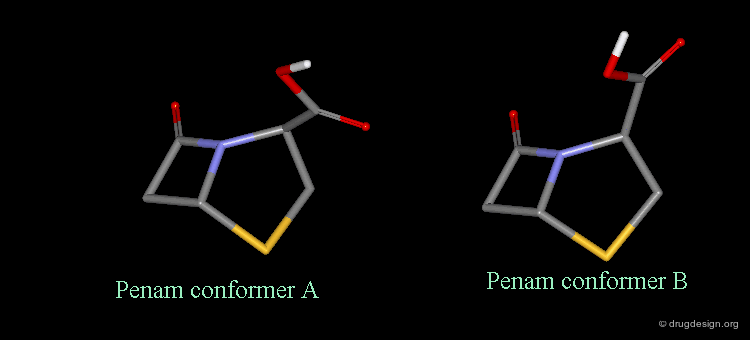
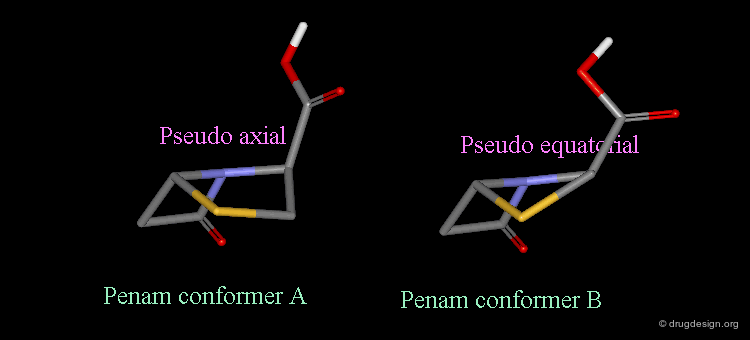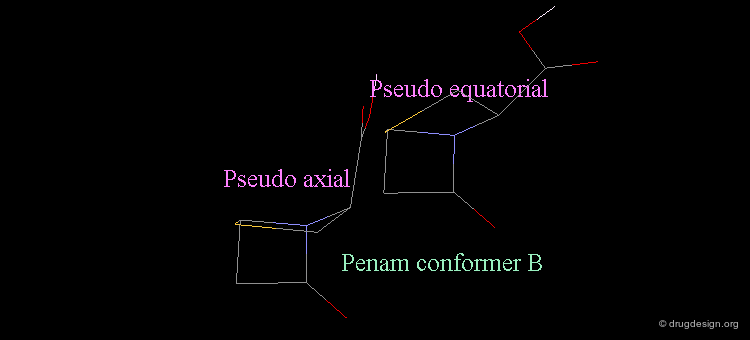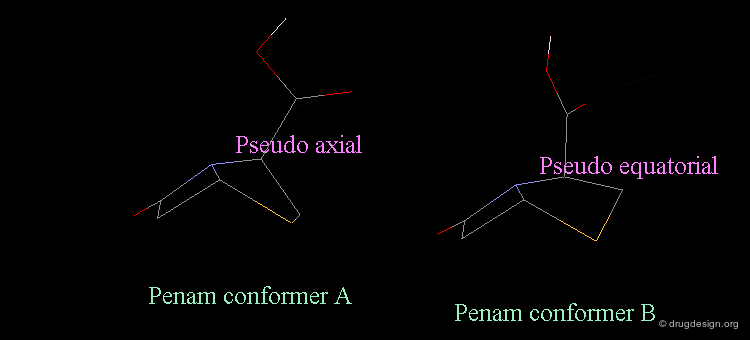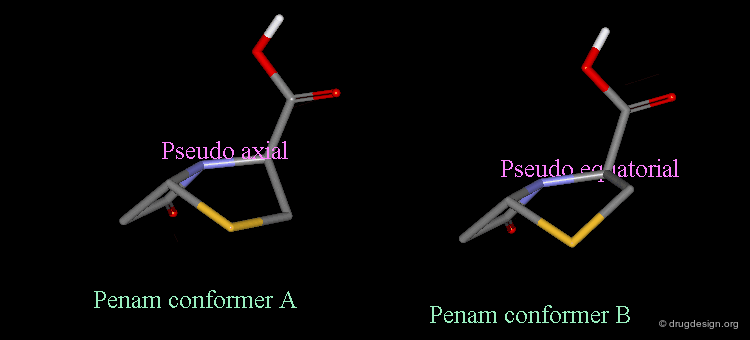
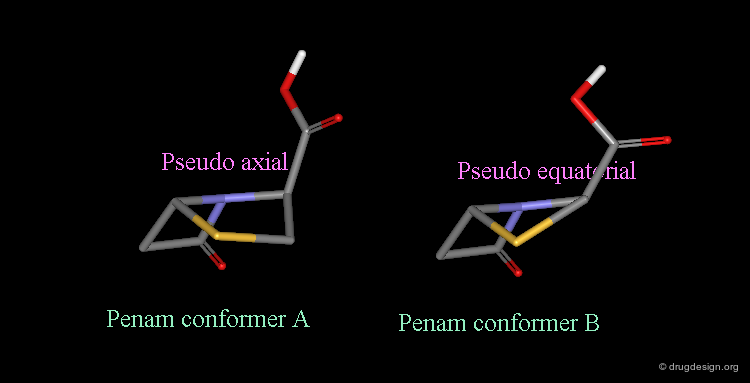
Pseudorotation of the Penam Ring¶
In the conformation shown on the left side, the carboxy group is in a pseudo-axial orientation (the X-ray data that was available and used in the attempts that failed to find a geometrical interpretation), whereas in the conformation shown on the right side it is pseudo-equatorial.
The two Forms were Found in X-ray Determinations¶
The two forms have been experimentally observed in crystallographic studies. Small variations of energies accompany this process, and the carboxy group can easily fluctuate between the two positions. Molecular modeling studies show that the pseudo-axial form is more stable than the pseudo-equatorial one (difference of energy of about 2.9 kJ/mol).
articles
Beta-Lactam Antibiotics: Geometrical Requirements for Antibacterial Activities Cohen NC J.Med.Chem. 28 1983
Revealing Bioactive Conformation of Penicillins¶
It is possible to establish a geometric separation between active and inactive structures by considering the penicillin in the pseudo-equatorial orientation. When the different structures are superimposed, the view reveals the sharp geometrical difference between the active and inactive structures.

Modeling Analyses¶
The initial difficulty in clarifying the 3D features was in fact due to the existence of a stable pseudo-axial form of penicillin. It appeared in molecular models and also in X-ray crystallography data as perfectly capable of mimicking the inactive compounds.
articles
Beta-Lactam Antibiotics: Geometrical Requirements for Antibacterial Activities Cohen NC J.Med.Chem. 28 1983
Separation Between Active and Inactive Molecules¶
As shown in the previous view, the active molecules have the carboxy group much closer to the β-lactam amide function than the three inactive geometries. The distance between the oxygen β-lactam amide and the carbon carboxy group was found to be a measure of activity. All active compounds have a distance of 3.0-3.9 Å, whereas inactive compounds have a distance of more than 4.1 Å.

articles
Beta-Lactam Antibiotics: Geometrical Requirements for Antibacterial Activities Cohen NC J.Med.Chem. 28 1983
Other Beta-Lactam Antibiotic Structures¶
Other active β-lactam antibiotic structures such as those indicated here were shown to fit well with the geometrical requirements analyzed here. For example the distance previously defined is 3.024 Å for the oxa-analog and 3.355 Å for Sulfazecin (oxygen-sulfur distance).

articles
Beta-Lactam Antibiotics: Geometrical Requirements for Antibacterial Activities Cohen NC J.Med.Chem. 28 1983
Browser of Beta-Lactam Antibiotics¶

Validation and Perspectives¶
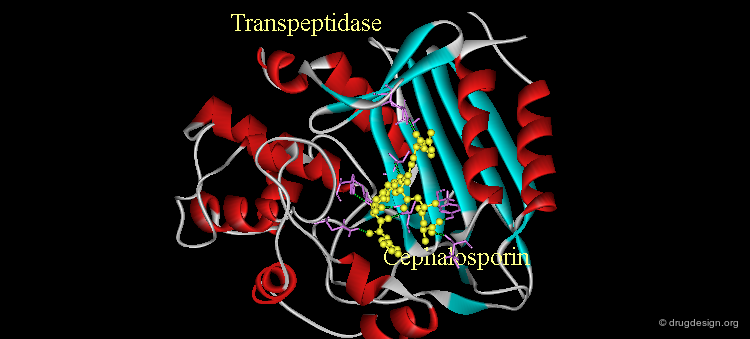
Attempts to restore antibiotic activities to the inactive Δ2 cephalosporin scaffold were not successful. The advent of X-ray crystallography of bacterial trans-peptidases has given new impetus to this area. These enzymes catalyze the formation of the cross-linked bacteria cell wall, a construction that provides structural strength and protection to bacteria. Here is shown the structure of a cephalosporin bound to the active site of a transpeptidase.
articles
A 1.2-A Snapshot of the Final Step of Bacterial Cell Wall Biosynthesis. Lee W, McDonough MA, Kotra L, Li ZH, Silvaggi NR, Takeda Y, Kelly JA,and Mobashery S Proc. Natl. Acad. Sci. USA. 98 2001
Case Study-7 : Anilino-Quinazoline Example¶
Potent Inhibitor of the EGF-R Protein Kinase¶
The following 4-anilino-quinazoline is a potent inhibitor of the EGF-R protein kinase. In the following pages we describe how structure-activity relationship analyses make it possible to pinpoint the molecular determinants that are responsible for the biological activity of this molecule.

articles
Chemical Inhibitors of Protein Kinases Bridges AJ Chem. Rev 101 2001
SAR: Substitution of Anilino N4¶
The N-methyl analog of the 4-anilino-quinazoline lead was prepared and proved to have very poor inhibitory activity.

articles
Chemical Inhibitors of Protein Kinases Bridges AJ Chem. Rev 101 2001
N-Methyl Analog¶
By comparing the initial anilino-quinazoline lead and its N-methyl analog, it might seem that the activity of the initial lead is due to the formation of a hydrogen bond between a carbonyl of the kinase and the N-H group of the lead.

articles
Chemical Inhibitors of Protein Kinases Bridges AJ Chem. Rev 101 2001
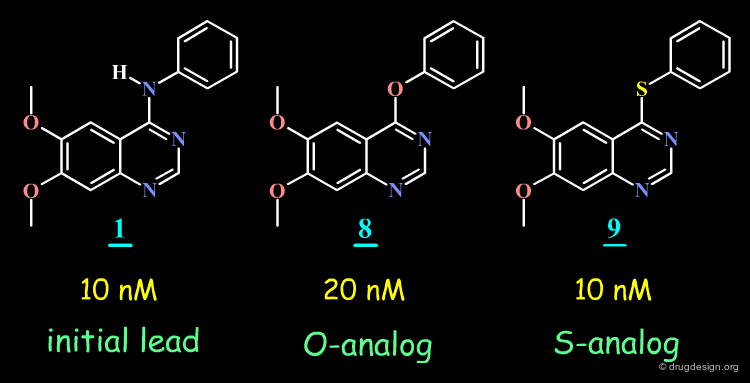
SAR Observations in a 4-Anilino-Quinazolines Series¶
The hypothesis put forward on the previous page does not hold since the O- and the S-analogs of the initial lead proved to be potent EGF-R kinase inhibitors.
articles
Chemical Inhibitors of Protein Kinases Bridges AJ Chem. Rev 101 2001
The preparation and SAR of 4-(anilino), 4-(phenoxy) and 4-(thiophenoxy)-quinazolines: inhibitors of p56-lck and EGF-R tyrosine kinase activity Myers MR, Setzer NN, Spada AP, Zulli AL, Hsu CYJ, Zilberstein A, Jhonson SE, Hook LE, Jacoski MV. Bioorg. and Med. Chem. Lett 7(4) 1997 None
Epidermal growth factor receptor throsine kinase: structure-activity relationships and antitumour activity of novel quinazolines Gibson KH, Grundy W, Godfrey AA, Woodburn JR, Ashton SE, Curry BJ, Scarlett L, Barker AJ, Brown DS. Bioorg. and Med. Chem. Lett 7(21) 1997 None
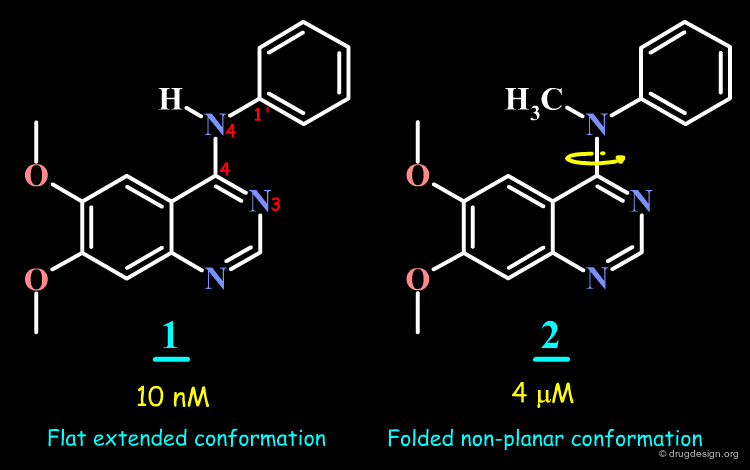
Conformation of 4-Anilino-Quinazoline Molecules¶
Conformational analyses indicate that the 4-anilino-quinazoline structure exists in an extended form, whereas in the N-Methyl analog, the aniline moiety cannot adopt an extended conformation because of steric repulsions.


Geometry of 4-Anilino-Quinazoline Structures¶
The analyses presented in the previous pages raise the possibility that the molecular geometry of the molecules is an important determinant for the biological activities of the 4-anilino-quinazoline molecules. This geometry is controlled by the following two torsion angles: Φ-1 and Φ-2.

articles
Chemical Inhibitors of Protein Kinases Bridges AJ Chem. Rev 101 2001
Experimental Conformations of Anilino-Quinazolines¶
A preliminary analysis of the conformation of free and bound anilino-quinazolines in the solid state and in solution shows that in general the molecules adopt an extended conformation (in parenthesis are indicated the values of the two torsion angles Φ-1 and Φ-2).

articles
Chemical Inhibitors of Protein Kinases Bridges AJ Chem. Rev 101 2001
4-Anilino-6,7-dialkoxyquinoline-3-carbonitrile inhibitors of epidermal growth factor receptor kinase and their bioisosteric relationship to the 4-anilino-6,7-dialkoxyquinazoline inhibitors Wissner A, Berger DM, Boschelli DH, Floyd MB Jr, Greenberger LM, Gruber BC, Johnson BD, Mamuya N, Nilakantan R, Reich MF, Shen R, Tsou HR, Upeslacis E, Wang YF, Wu B, Ye F, Zhang N. "J. Med. Chem 43(17) 2000 None
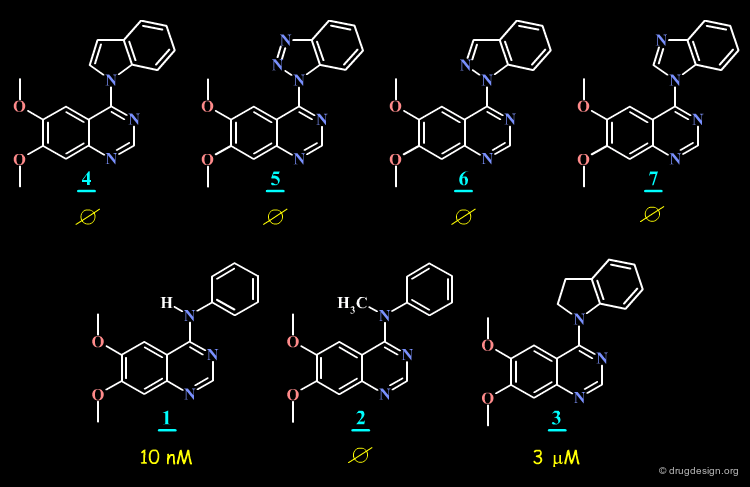
SAR: Substitution of N4 is Detrimental to Potency¶
Further SAR analyses indicate that the substitution of N4 in 4-anilino-quinazolines is detrimental to their biological activities.
articles
Chemical Inhibitors of Protein Kinases Bridges AJ Chem. Rev 101 2001
The preparation and SAR of 4-(anilino), 4-(phenoxy) and 4-(thiophenoxy)-quinazolines: inhibitors of p56-lck and EGF-R tyrosine kinase activity Myers MR, Setzer NN, Spada AP, Zulli AL, Hsu CYJ, Zilberstein A, Jhonson SE, Hook LE, Jacoski MV. Bioorg. and Med. Chem. Lett 7(4) 1997 None
Epidermal growth factor receptor throsine kinase: structure-activity relationships and antitumour activity of novel quinazolines Gibson KH, Grundy W, Godfrey AA, Woodburn JR, Ashton SE, Curry BJ, Scarlett L, Barker AJ, Brown DS. Bioorg. and Med. Chem. Lett 7(21) 1997 None
Torsion Angle N3-C4-N4-C1' is Important for Potency¶
The SAR data assembled for the 4-anilino-quinazoline suggest that the torsion angle Φ (N3-C4-N4-C1') which controls the position of the anilino phenyl group is important for potency; since good biological activities are associated with small values for this angle (close to zero degrees), and correspond to a flat structure with an extended conformation.
articles
Chemical Inhibitors of Protein Kinases Bridges AJ Chem. Rev 101 2001
Energy of Bioactive Conformers¶
Analysis of the energy of the conformation of the molecules in their extended conformation indicate that this form corresponds to the conformation of lowest energy for molecules 1 (initial lead), 8 and 9 (O- and S- analogs) but that this form corresponds to high energies for molecules 2, 4-7 and 3.

articles
Unpublished N. C. Cohen None None 2002
Browser of Selected Anilino-Quinazoline Analogs¶
The SAR analyses of the 4-anilino-quinazoline structures discussed here leads to the conclusion that the extended conformation corresponds to the bioactive conformation of the molecules, with the anilino moiety close to coplanarity with the plane of the quinazoline nucleus.

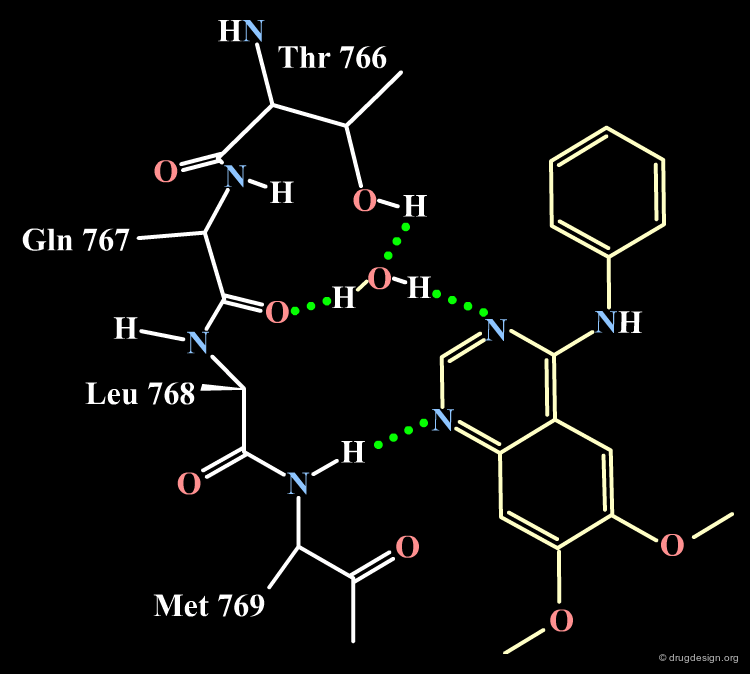
Docking of 4-Anilino-Quinazoline Lead¶
The docking of the 4-anilino-quinazoline lead with the EGF-R protein kinase reveals the binding mode described in the following figures. Note that the N-H anilino group is not involved in the formation of hydrogen bonds with the receptor; note also that the two substituents at positions 7 and 9 are pointing towards the outside of the protein (a feature that will be exploited for conceiving more soluble drug candidates). These predictions were confirmed by X-ray studies.

articles
Chemical Inhibitors of Protein Kinases Bridges AJ Chem. Rev 101 2001
Summary of Structural Analyses¶
A summary of the structural determinants deduced by SAR analyses of the 4-anilino-quinazoline analogs is given below.

articles
Chemical Inhibitors of Protein Kinases Bridges AJ Chem. Rev 101 2001
ADDITIONAL CASE STUDIES¶
Additional Case Studies¶

Copyright © 2024 drugdesign.org