Structure Activity Relationships¶
Info
When a new type of active compound is discovered, analogs are investigated to gain insights into its structure and to discover useful drug candidates. SAR analyses try to guide this process by converting structure-activity results into informative structure-activity relationships in molecular terms, and to ensure a good exploitation of this knowledge. This chapter presents the concepts underlying SAR and illustrates them with concrete examples.
Number of Pages: 157 (±3 hours read)
Last Modified: May 2005
Prerequisites: None
Introduction¶
Structure Activity Relationships (SAR)¶
When a new type of active compound is discovered the chemist through alterations in its molecular structure, attempts to produce new compounds with similar, perhaps improved activity, or with other valuable active properties. At a given stage the effects observed are interpreted in terms of structural variations, which are then used to decide which modifications to consider next. Relating biological activities to molecular structures is known as structure-activity relationships (SAR).

Aim of SAR Analyses¶
SAR analyses try to convert structure-activity observations into informative structure-activity relationships. They aim at maximizing the knowledge that can be extracted from the raw data in molecular terms, exploit this knowledge to identify which molecules should be synthesized and identify lead compounds for either additional modifications or further pre-clinical studies.
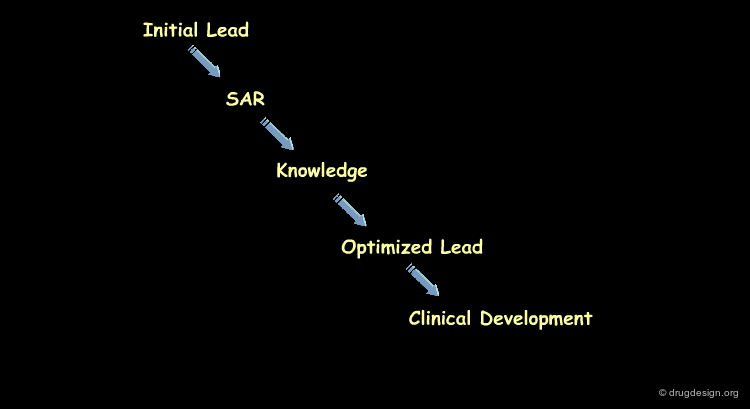
Results of a SAR Analysis¶
The results of a SAR analysis can be represented in a condensed form, either in a ligand-based or a receptor-based approach, as illustrated below. They represent substantial amounts of time and money invested in human resources, scientific effort and human creativity in the project concerned.
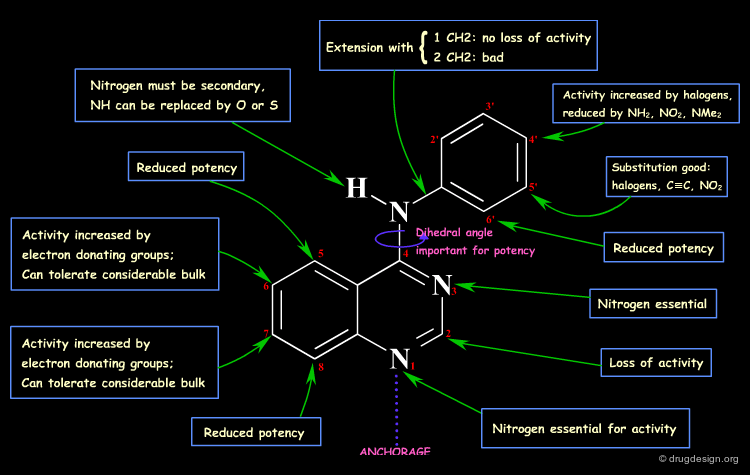

articles
Chemical Inhibitors of Protein Kinases Bridges AJ Chem. Rev 101 2001
Principle: Alteration of an Active Substance¶
The progressive alteration of the molecular structure of a reference compound allows for the determination of the importance of the structural elements involved. This can be done by removing, adding or replacing specific molecular fragments and testing how the structural variation affects biological activities. For example, if the analog is inactive, the original functional group is interpreted as being essential to binding; if the activity is not affected, the original functional group is considered to be unimportant. Each of the novel molecules synthesized is expected to yield useful knowledge.
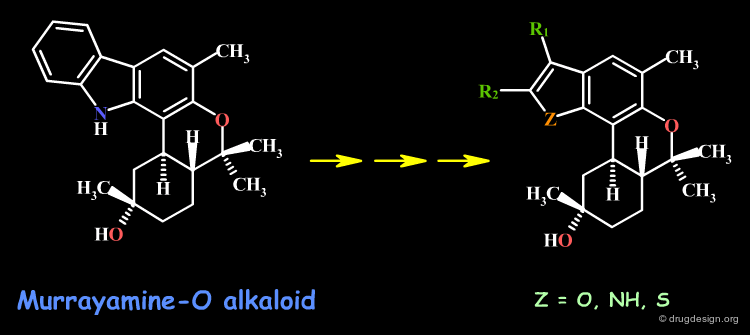
articles
Murrayamine-O and -P, Two Cannabinol-skeletal Carbazole Alkaloids from Murraya euchrestifolia Wu TS, Wang ML and Wu PL Tetrahedron Letters 36 1995
Development: a Single Modification at a Time¶
Data of high informational content can be obtained when derived from single structural modifications of an initial lead structure. The introduction of multiple changes should be avoided because of the difficulties created for the correct interpretation of the biological results. The example below illustrates a molecule for which three alterations were introduced. In the absence of other SAR, the interpretation of the inactivity of the resulting molecule is impossible.

Iterative Process¶
The approach proceeds with successive iterations. It starts with the analysis of the structure of the initial target molecule. Many questions are raised, for which the chemist can identify candidate molecules that may be able to provide answers to the questions. In parallel, he looks for a synthetic program that can generate valuable active candidates to be further fine-tuned in the subsequent loops.

Chemical Modifications and Medicinal Chemist Tools¶
The tools that are available to the medicinal chemist are diverse. Depending on the structural fragments that captured his interest during the analyses, the synthetic program can involve chemical modifications such as those described below.

Chemistry is the Limiting Factor¶
The implementation of the strategy is however biased by the actual synthetic possibilities; for example the systematic variation of all kind of substitutions in a ring is not always feasible. An intelligent program that allows for multiple variations in the final stage of the syntheses, although desirable is still biased, and covers only a small portion of the entire space of possible modifications.
Role of the Functional Groups in the Reference Structure¶
In current practice, many of the modifications that are implemented by medicinal chemists involve the functional groups of the reference compound. The systematic alteration of selected functional groups serves to probing their possible interactions with the receptor and to test their relevance to binding in terms of hydrogen bonds, hydrophobic or polar interactions. This will be presented in the following pages.

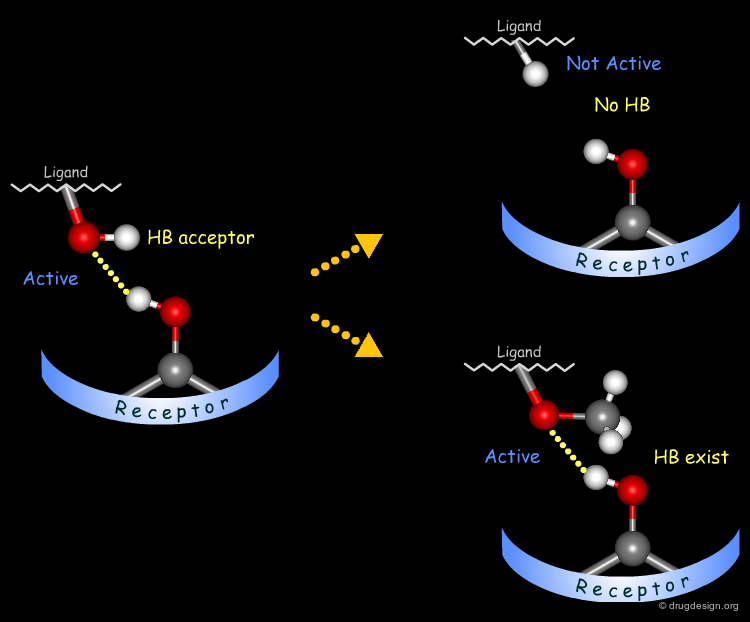
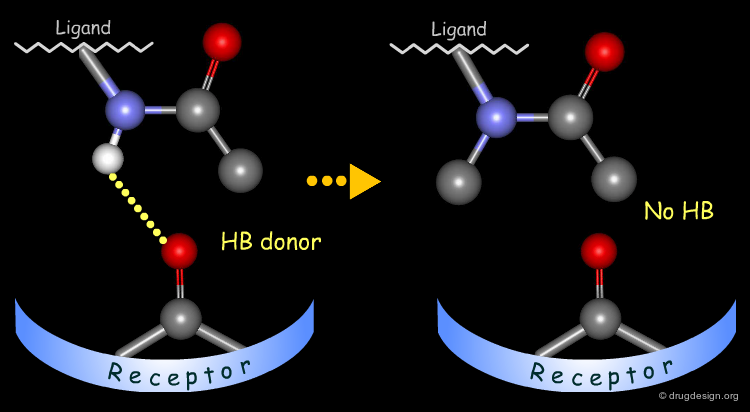
Probing H-Bond Interactions¶
Principle for Probing Hydrogen Bond Interactions¶
The method for probing the existence of hydrogen bond interactions with a specific functional group consists of preparing analogs where the functional group concerned is unable to make hydrogen bonds.

Hydroxyl: Hypothetical H-Bond Interactions¶
Hydroxyl groups of alcohols, phenols, enols, oximes etc... can act as hydrogen bond donors or acceptors.

Testing the Existence of H-Bond Interactions¶
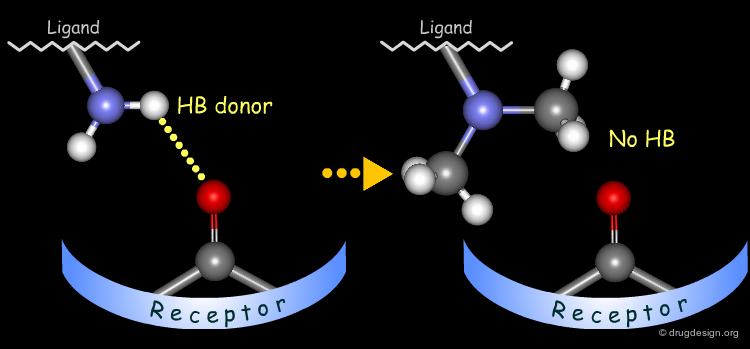
Simple alterations of hydroxyl functions can be made to disrupt existing hydrogen bonds, as for example replacing the OH by H or by CH3.

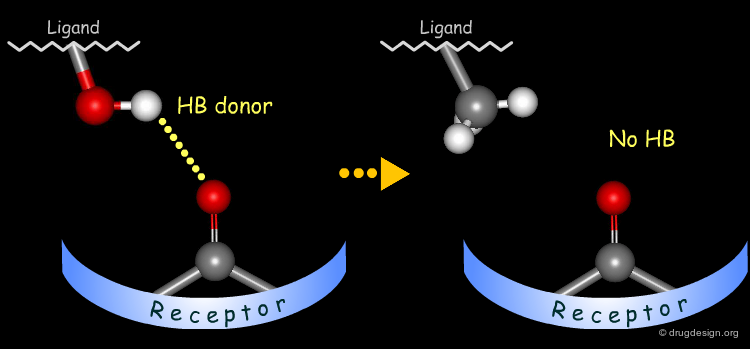
Testing H-Bond Donor Capability of the Hydroxyl¶
To test whether a hydroxyl group is involved as a donor of hydrogen bonds, it is possible to alkylate the hydroxyl or to prepare the corresponding ketone. If the biological activity drops, one can conclude that this hydroxyl group is important and is likely to interact with the protein as a hydrogen bond donor.

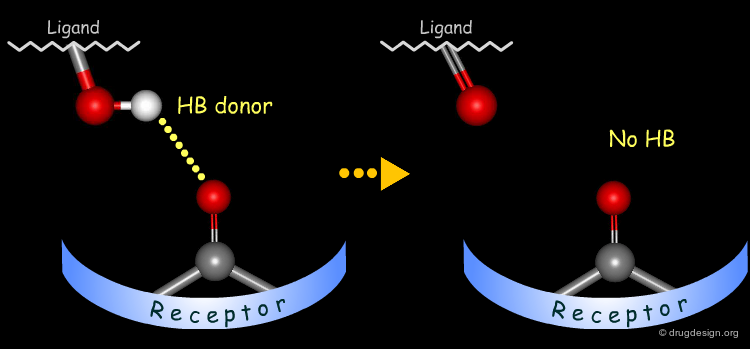
Testing H-Bond Acceptor Capability of the Hydroxyl¶
It is difficult to simply test whether a hydroxyl group is involved as an acceptor of hydrogen bonds: no simple alterations can be envisaged that would disrupt the existing hydrogen bond (both the methoxy and the C=O analogs can still have their oxygen atom involved as a hydrogen bond acceptor).

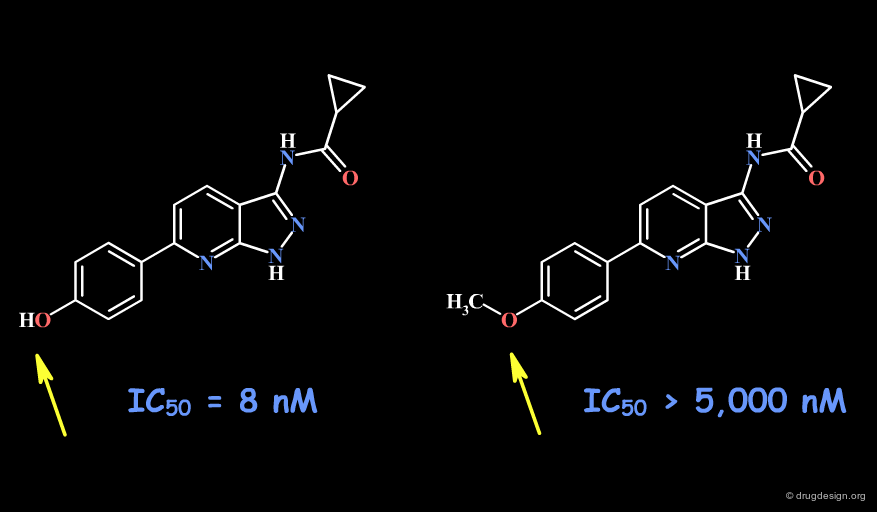
Example 1: Pyrazolopyrimidines¶
In the following example, replacing the phenolic OH by a methoxy, leads to the complete loss of biological activity. The hydroxyl is likely to form a hydrogen bond with the receptor (as a hydrogen bond donor). Other factors including changes in the volume, the physico-chemical properties of the molecule such as the polar surface area, acidity, hydrophobicity, solubility, solvation etc... must also be envisaged.
articles
6-Aryl-pyrazolo[3,4-b]pyridines: Potent Inhibitors of Glycogen Synthase Kinase-3 (GSK-3) Witherington J, Bordas V, Gaiba A, Garton NS, Naylor A, Rawlings AD, Slingsby BP, Smith DG, Takle AK and Ward RW Bioorganic and Medicinal Chemistry Letters 13 2003
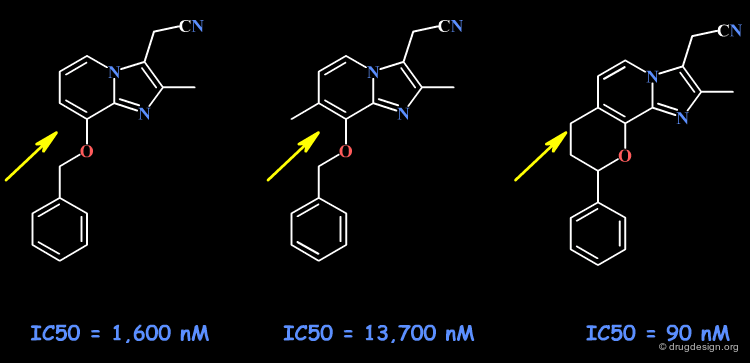
Example 2: Benzimidazoles¶
In the example here, the phenolic OH was replaced by both a methoxy and a hydrogen atom. The phenolic hydroxyl is likely to form a hydrogen bond with the receptor (as a hydrogen bond donor). Other factors including changes in the volume, the physico-chemical properties of the molecule such as the polar surface area, acidity, hydrophobicity, solubility, solvation etc... must also be envisaged.

articles
NR2B-Selective N-Methyl-D-aspartate Antagonists: Synthesis and Evaluation of 5-Substituted Benzimidazoles McCauley JA, Theberge CR, Romano JJ, Billings SB, Anderson KD, Claremon DA, Freidinger RM, Bednar RA, Mosser SD, Gaul SL, Connolly TM, Condra CL, Xia M, Cunningham ME, Bednar B, Stump GL, Lynch JJ, Macaulay A, Wafford KA, Koblan KS and Liverton NJ J. Med. Chem 47 2004
Example 3: Pyrrolopyrimidine¶
Replacing the phenolic OH by a methoxy, leads to a drop in biological activity. The hydroxyl may form a hydrogen bond with the receptor (as a hydrogen bond donor). Other factors including changes in the volume, the physico-chemical properties of the molecule such as the polar surface area, acidity, hydrophobicity, solubility, solvation etc... must also be envisaged.

articles
7-Alkyl- and 7-Cycloalkyl-5-aryl-pyrrolo[2,3-d ]pyrimidines. Potent Inhibitors of the Tyrosine Kinase c-Src Widler L, Green J, Missbach M, Susa M and Altmann E Bioorganic and Medicinal Chemistry Letters 11 2001
Example 4: Salicylanilides¶
The replacement of the phenolic OH by a methoxy, leads to the complete loss of the biological activity: the hydroxyl is therefore important and is likely be involved as a hydrogen bond donor. But is it an intra-molecular hydrogen bond (fixing the conformation in a bioactive geometry) or a hydrogen bond that is formed with the receptor? Other factors including changes in the volume, the physico-chemical properties of the molecule such as the polar surface area, acidity, hydrophobicity, solubility, solvation etc... must also be envisaged.

articles
Salicylanilides as inhibitors of the protein tyrosine kinase epidermal growth factor receptor Liechti C, Sequin U, Bold G, Furet P, Meyer T and Traxler P European Journal of Medicinal Chemistry 39 2004
Example 5: Isoflavones¶
In this example the replacement of the phenolic hydroxyls yields a completely inactive molecule. The two hydroxyl groups may be involved as hydrogen bond donors. Other factors including changes in the volume, the physico-chemical properties of the molecule such as the polar surface area, acidity, hydrophobicity, solubility, solvation etc... must also be envisaged.

articles
Use of a Pharmacophore Model for the Design of EGFR Tyrosine Kinase Inhibitors: Isoflavones and 3-Phenyl-4(1H)-quinolones Traxler P, Green J, Mett H, Sequin U and Furet P Journal of Medicinal Chemistry 42 1999
Carbonyl: Hypothetical H-Bond Interactions¶
A carbonyl group can act only as a hydrogen bond acceptor.

Testing the Existence of H-Bond Interactions¶
To test whether a carbonyl group is involved as an acceptor of hydrogen bonds, it is possible to replace the C=O by a C=CH2, a CH2 or reduce it to an alcohol. If the biological activity drops, one can conclude that this carbonyl group may interact with the protein as a hydrogen bond acceptor.



Example 1: Aminobenzophenones¶
The SAR indicate that the carbonyl group of this series may form a hydrogen bond with the receptor site. Other changes including the volume, the conformational and the physico-chemical properties of the molecules such as the polar surface area, hydrophobicity, solubility, solvation etc... must also be envisaged.

articles
Synthesis and Structure-Activity Relationship of Aminobenzophenones. A Novel Class of p38 MAP Kinase Inhibitors with High Antiinflammatory Activity Ottosen ER, Sorensen MD, Bjorkling F, Skak-Nielsen T, Fjording MS, Aaes H and Binderup L J. Med. Chem 46 2003
Example 2: Thiazolidine-dione¶
The carbonyl group may form a hydrogen bond with the receptor site. Other changes including the volume, the conformational and the physico-chemical properties of the molecules such as the polar surface area, hydrophobicity, solubility, solvation etc... must also be envisaged.
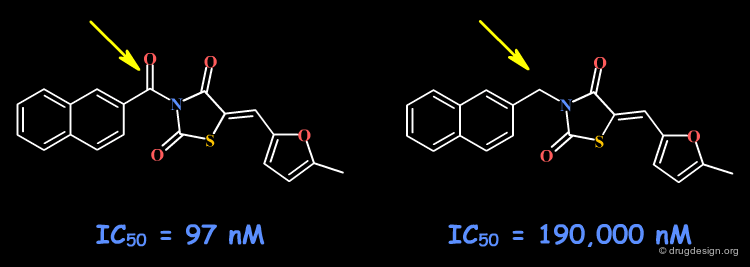
articles
Identification of a Stable Chymase Inhibitor Using a Pharmacophore-Based Database Search Koide Y, Tatsui A, Hasegawa T, Murakami A, Satoh S, Yamada H, Kazayamaa SI and Takahashia A Bioorganic and Medicinal Chemistry Letters 13 2003
Example 3: Pyrazolopyridines¶
The SAR observed for the three analogs synthesized indicate that the C=O may form a hydrogen bond with the receptor site. Other changes including the volume, the conformational and the physico-chemical properties of the molecules such as the polar surface area, hydrophobicity, solubility, solvation etc... must also be envisaged.

articles
1H-Pyrazolo[3,4-b]pyridine Inhibitors of Cyclin-Dependent Kinases Misra RM, Rawlins DB, Xiao HY, Shan W, Bursuker I, Kellar KA, Mulheron JG, Sack JS, Tokarski JS, Kimball SD and Webster KR Bioorganic and Medicinal Chemistry Letters 13 2003
Example 4: Naphthyl Ketones¶
The C=O group may form a hydrogen bond with the receptor site. Other changes including the volume, the conformational and the physico-chemical properties of the molecules such as the polar surface area, hydrophobicity, solubility, solvation etc... must also be envisaged.
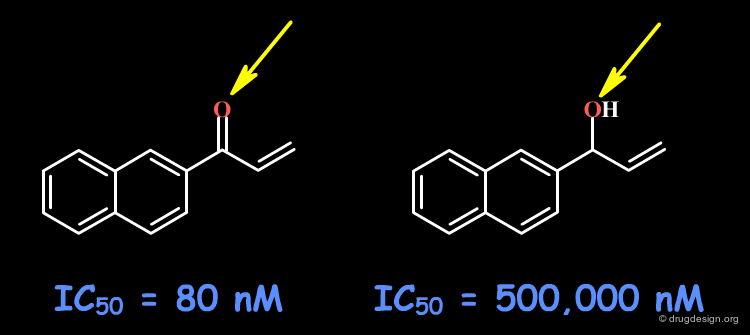
articles
Naphthyl Ketones: A New Class of Janus Kinase 3 Inhibitors Brown GR, Bamford A, Bowyer J, James DS, Rankine N, Tang E, Torr V and Culbert EJ Bioorganic and Medicinal Chemistry Letters 10 2000
Example 5: Cyclic Peptides¶
The carbonyl group may form a hydrogen bond with the receptor site (an internal hydrogen bond is excluded because of no hydrogen bond donor candidate in the peptide molecule considered). Other changes including the volume, the conformational and the physico-chemical properties of the molecule such as the polar surface area, hydrophobicity, solubility, solvation etc... must also be envisaged.

book
Freidinger RM, Bock MG, Clineschmidt BV, DiPardo RM, Erb JM, Evans BE, Gilbert KF, Gould NP, Hobbs DW, Hoffman JB, Leighton JL, Lundell GF, Perlow DS, Pettibone DJ, Tung RD, Veber DF, Whitter WL and Williams PD Prespectives in Medicinal Chemistry Verlag Helvetica Chimica Acra, Basel 1993
Amide¶
Amides are capable of acting as hydrogen bond donors (with the N-H) or acceptors (with either the nitrogen atom of the N-H or the oxygen atom of the C=O).

Testing the Existence of H-Bond Interactions¶
Testing the existence of hydrogen bond interactions of an amide group can be made by replacing this fragment by a trans double-bond.

Testing H-Bond Acceptor Capability of the Carbonyl¶
It is possible to probe the hydrogen bond capability of the carbonyl of an amide fragment by making the corresponding ene-amine.

Testing H-Bond Donor Capability of the Nitrogen¶
It is possible to test the hydrogen bond donor capability of the N-H group of an amide group by replacing the N-H by a N-CH3.
Example 1: Lactam Tricylic¶
The SAR indicate that the N-H group may be involved as a hydrogen bond donor and the carbonyl oxygen as a hydrogen bond acceptor. Other changes including the volume, the conformational and the physico-chemical properties of the molecules such as the polar surface area, pKa, hydrophobicity, solubility, solvation etc... must also be envisaged.

articles
Novel Tricyclic Poly(ADP-ribose) Polymerase-1 Inhibitors with Potent Anticancer Chemopotentiating Activity: Design, Synthesis, and X-ray Cocrystal Structure Koch SSC, Thoresen LH, Tikhe JG, Maegley KA, Almassy RJ, Li J, Yu XH, Zook SE, Kumpf RA, Zhang C, Boritzki TJ, Mansour RN, Zhang KE, Ekker A, Calabrese CR, Curtin NJ, Kyle S, Thomas HD, Wang LZ, Calvert AH, Golding BT, Griffin RJ, Newell DR, Webber SE and Hostomsky Z J. Med. Chem 45 2002
Example 2: Pyrrolopyrimidinones¶
The replacement of the hydrogen atom by a methyl indicates that the N-H group may be involved as a hydrogen bond donor in the interaction of the original molecule with the receptor site. Other changes including the volume, the conformational and the physico-chemical properties of the molecules such as the polar surface area, pKa, hydrophobicity, solubility, solvation etc... must also be envisaged.

articles
Rational Design of 4,5-Disubstituted-5,7-dihydro-pyrrolo[2,3-d]pyrimidin-6-ones as a Novel Class of Inhibitors of Epidermal Growth Factor Receptor (EGF-R) and Her2(p185erbB) Tyrosine Kinases Sun L, Cui J, Liang C, Zhou Y, Nematalla A, Wang X, Chen H, Tang C and Wei J Bioorganic and Medicinal Chemistry Letters 12 2002
Primary Amines¶
Primary amines are capable of acting as hydrogen bond donors or acceptors.

Testing the Existence of H-Bond Interactions¶
Simple alterations of primary amine functions can be made to disrupt existing hydrogen bonds, as for example replacing the NH2 by H or by CH3.


Testing H-Bond Donor Capability of the Amines¶
It is possible to test the hydrogen bond capability of a primary amine group by replacing the NH2 by N(CH3)2.
Example 1: Imidazole Acetic Acids¶
The SAR data obtained in this example indicate that the NH2 group may be involved with two hydrogen bonds in the interaction of the molecule with the receptor. Other changes including the geometrical and the physico-chemical properties of the molecules such as the polar surface area, pKa, hydrophobicity, solubility, solvation etc... must also be envisaged.

articles
Imidazole acetic acid TAFIa inhibitors: SAR studies centered around the basic P'1 group Nantermet PG, Barrow JC, Lindsley SR, Young M, Mao SS, Carroll S, Bailey C, Bosserman M, Colussi D, McMasters DR, Vaccaa JP and Selnick HG Bioorganic and Medicinal Chemistry Letters 14 2004
Example 2: Salicylanilides¶
The amino group may be involved as a hydrogen bond donor in the interaction with the receptor site. Other changes including the volume, the geometrical and the physico-chemical properties of the molecules such as the polar surface area, pKa, hydrophobicity, solubility, solvation etc... must also be envisaged.
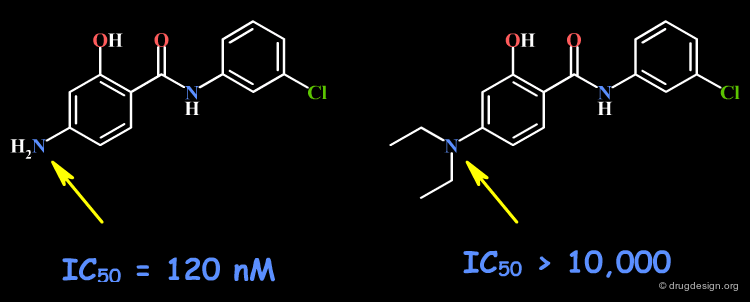
articles
Salicylanilides as inhibitors of the protein tyrosine kinase epidermal growth factor receptor Liechti C, Sequin U, Bold G, Furet P, Meyer T and Traxler P European Journal of Medicinal Chemistry 39 2004
Secondary Amines¶
Secondary amines are capable of acting as hydrogen bond donors or acceptors.

Testing the Existence of H-Bond Interactions¶
To test for the existence of hydrogen bond interactions with a secondary amine can be made by replacing the N-H by a CH2 group.

Testing H-Bond Donor Capability of the Amines¶
To test for the hydrogen bond capability of a secondary amine can be made by replacing the N-H by N-CH3 or by O, S.



Example 1: Imidazoquinoxalines¶
The N-H group may act as a hydrogen bond donor in the interaction of the original molecule with the receptor. Other changes including the volume and the physico-chemical properties of the molecules such as the polar surface area, basicity, hydrophobicity, solubility, solvation etc... must also be envisaged.

articles
Discovery and Initial SAR of Imidazoquinoxalines as Inhibitors of the Src-Family Kinase p56Lck Chen P, Norris D, Iwanowicz EJ, Spergel SH, Lin J, Gu HH, Shen Z, Wityak J, Lin TA, Pang S, De Fex HF, Pitt S, Shen DR, Doweyko AM, Bassolino DA, Roberge JY, Poss MA, Chen BC, Schievend GL and Barrisha JC Bioorganic and Medicinal Chemistry Letters 12 2002
Example 2: Anilinopyrimidines¶
The N-H group may act as a hydrogen bond donor in the interaction with the receptor. Other changes including volume and the physico-chemical properties of the molecules such as the polar surface area, basicity, hydrophobicity, solubility, solvation etc... must also be envisaged.

articles
2-Anilino-4-(thiazol-5-yl)pyrimidine CDK inhibitors: synthesis, SAR analysis, X-ray crystallography, and biological activity Wang S, Meades C, Wood G, Osnowski A, Anderson S, Yuill R, Thomas M, Mezna M, Jackson W, Midgley C, Griffiths G, Fleming I, Green S, McNae I, Wu SY, McInnes C, Zheleva D, Walkinshaw MD, Fischer PM J. Med. Chem 47 2004
Example 3: Aminoquinazolines¶
By comparing the amino-quinazoline and its N-Methyl analog, one could think of a H-bond interaction with the N-H. This is not true, as evidenced by the O and S analogs that retain good activities. In the N-Methyl analog, the aniline moiety cannot adopt the extended bioactive conformation (steric repulsions), and this has been proven by X-ray crystallography (see next sub-page).
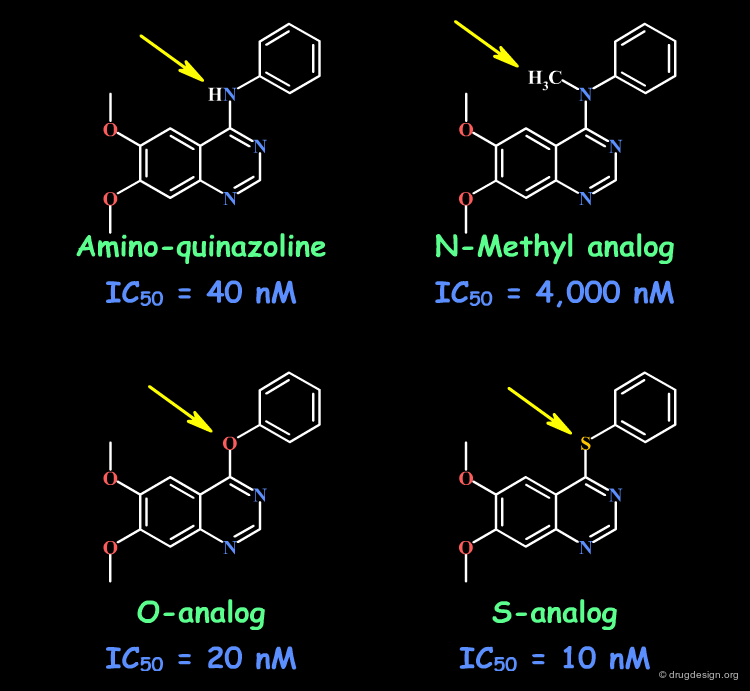
articles
Chemical Inhibitors of Protein Kinases Bridges AJ Chem. Rev 101 2001
Browser of Aminoquinazolines Analogs¶
Use this browser to compare different Aminoquinazoline Analogs in 3D.

Tertiary Amines¶
Tertiary amines can interact only as hydrogen bond acceptors.

Testing the Existence of H-Bond Interactions¶
It is possible to test for the existence of hydrogen bonds with a tertiary amine by making the analog where the nitrogen is replaced by a carbon atom.

Example Dihydropyridopyrimidones¶
As revealed by the SAR the nitrogen atom is important to binding. This atom may form a hydrogen bond with the receptor. Other changes including the volume and the physico-chemical properties of the molecules such as the polar surface area, basicity, hydrophobicity, solubility, solvation etc... must also be envisaged.

articles
p38MAP Kinase Inhibitors. Part 1: Design and Development of a New Class of Potent and Highly Selective Inhibitors Based on 3,4-Dihydropyrido[3,2-d]pyrimidone Scaffold Natarajan SR, Wisnoski DD, Singh SB, Stelmach JE, O'Neill EA, Schwartz CD, Thompson CM, Fitzgerald CE, O'Keefe SJ, Kumar S, Hop CECA, Zaller DM, Schmatzb DM and Dohertya JB Bioorganic and Medicinal Chemistry Letters 13 2003
Aromatic Nitrogens¶
A nitrogen atom on an aromatic ring can act as a hydrogen bond donor if the amine is secondary, or acceptor.

Testing the Existence of H-Bond Interactions¶
Testing the existence of hydrogen bond interactions with an aromatic nitrogen can be made by replacing the N-H by a C-H group.
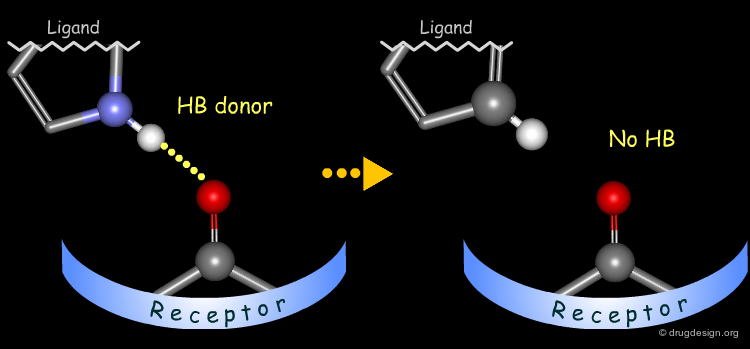
Testing H-Bond Donor Capability of the Amines¶
The hydrogen bond donor capability of an aromatic nitrogen can be tested by replacing the N-H by N-CH3 or by O,S.


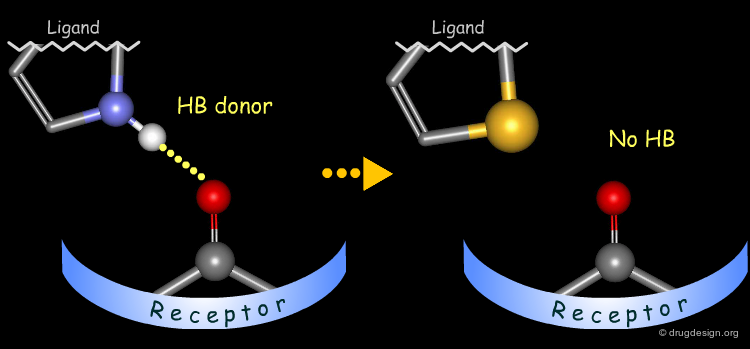
Example 1: Imidazoles and Oxazoles¶
The SAR in this example indicates that the N-H group may form a hydrogen bond with the receptor site. Other changes including the volume and physico-chemical properties of the molecules such as the polar surface area, basicity, hydrophobicity, solubility, solvation etc... must also be envisaged.

articles
SAR of 2,6-Diamino-3,5-difluoropyridinyl Substituted Heterocycles as Novel p38MAP Kinase Inhibitors Revesz L, Di Padova FE, Buhl T, Feifel R, Gram H, Hiestand P, Manning U, Wolf R and Zimmerlin AG Bioorganic and Medicinal Chemistry Letters 12 2002
Example 2: Pyrrolyl Ureas¶
The N-H group may form a hydrogen bond with the receptor. Other changes including the volume and physico-chemical properties of the molecules such as the polar surface area, basicity, hydrophobicity, solubility, solvation etc... must also be envisaged.

articles
p38 Kinase Inhibitors for the Treatment of Arthritis and Osteoporosis: Thienyl, Furyl, and Pyrrolyl Ureas Redman AM, Johnson JS, Dally R, Swartz S, Wild H, Paulsen H, Caringal Y, Gunn D, Renick J, Osterhout M, Kingery-Wood J, Smith RA, Lee W, Dumas J, Wilhelm SM, Housley TJ, Bhargava A, Ranges GE, Shrikhande A, Young D, Bombarab M and Scotta WJ Bioorganic and Medicinal Chemistry Letters 11 2001
Example 3: Anilinoquinazolines¶
Analogs of the 4-Anilino quinazoline structure were prepared to test the importance of the nitrogen atoms in this scaffold. The SAR indicate that a nitrogen atom needs to be present in positions 1 and 3, with however a greater importance for N1 as compared to N3 (compare the biological activities of B and C).
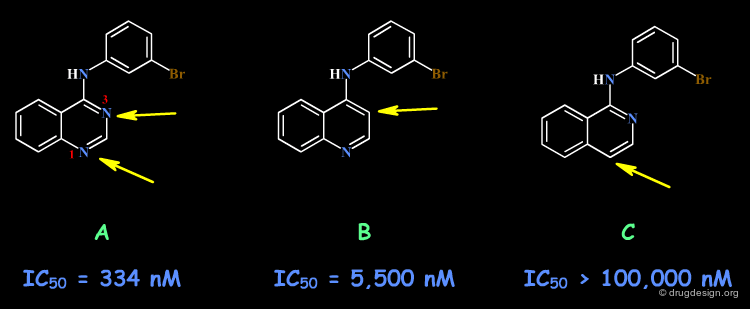
articles
Tyrosine kinase inhibitors. 5. Synthesis and structure-activity relationships for 4-[(phenylmethyl)amino]- and 4-(phenylamino)quinazolines as potent adenosine 5'-triphosphate binding site inhibitors of the tyrosine kinase domain of the epidermal growth factor receptor Rewcastle GW, Denny WA, Bridges AJ, Zhou H, Cody DR, McMichael A and Fry DW J. Med. Chem 38 1995
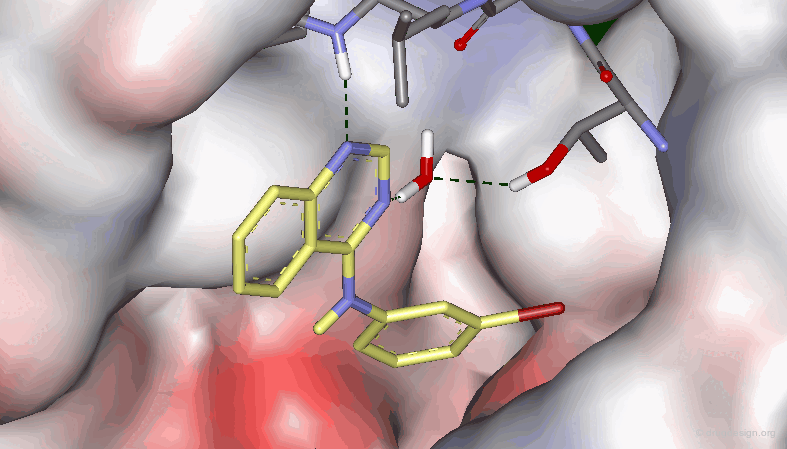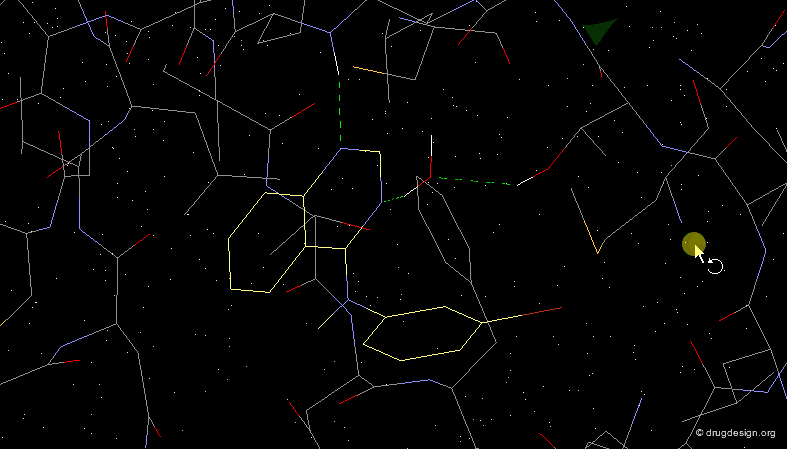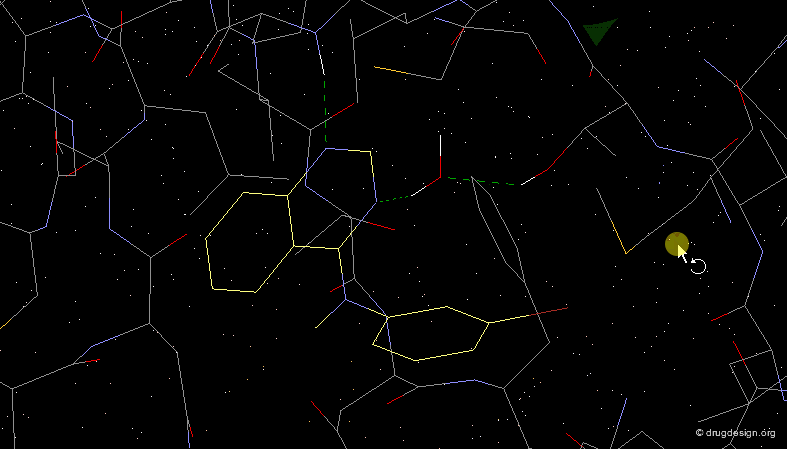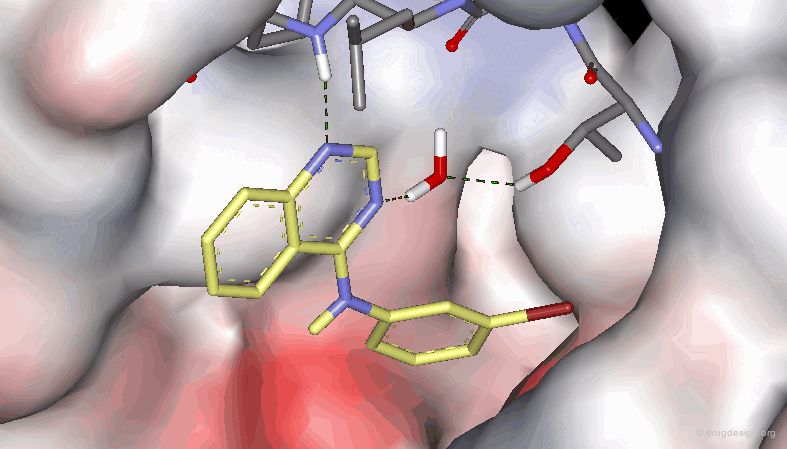
Example 3: X-Ray Data¶
Recent X-Ray studies of 4-Anilino quinazolines in complexes with the target protein revealed that the binding of the quinazoline scaffold involves hydrogen bond interactions with both N1 and N3: there is a direct hydrogen bond with N1, and an indirect one (via a water molecule) with N3.
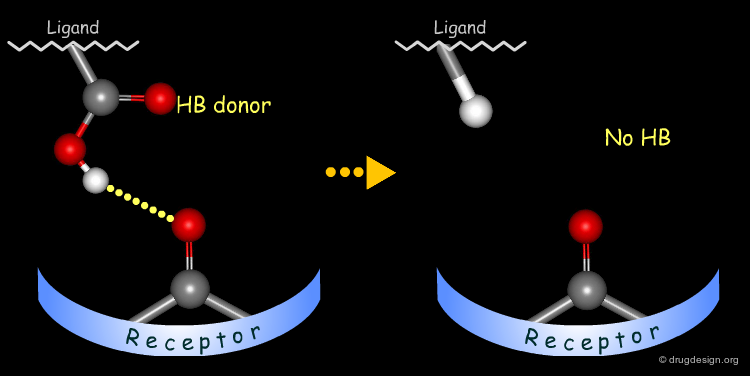
Carboxylic Acids¶
As far as hydrogen bonds are concerned, carboxylic acid groups can act as hydrogen bond donors or acceptors.
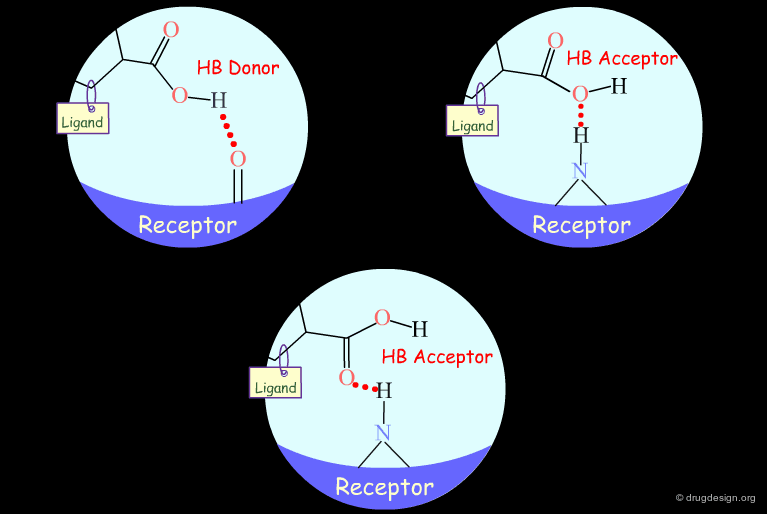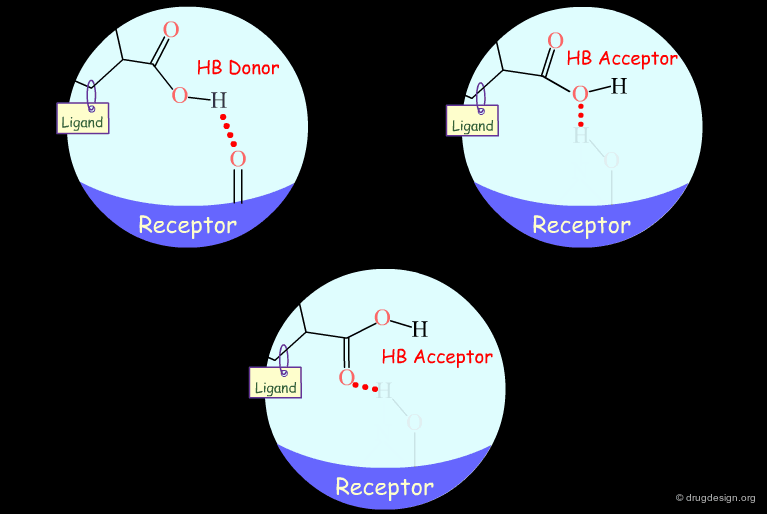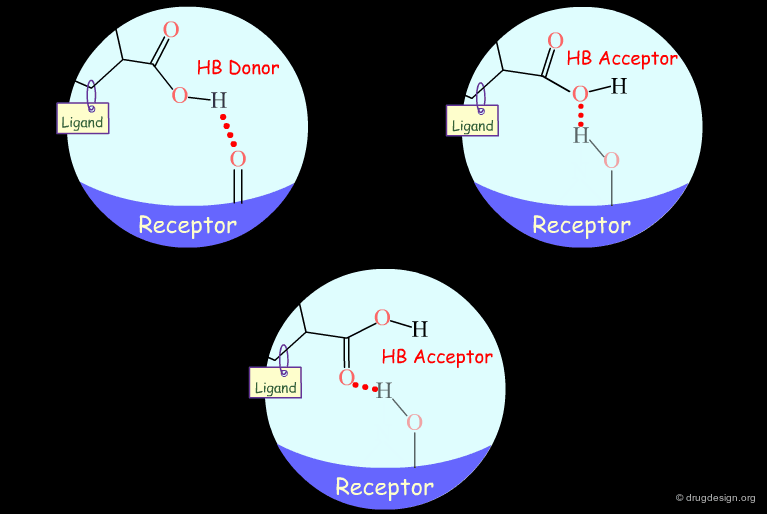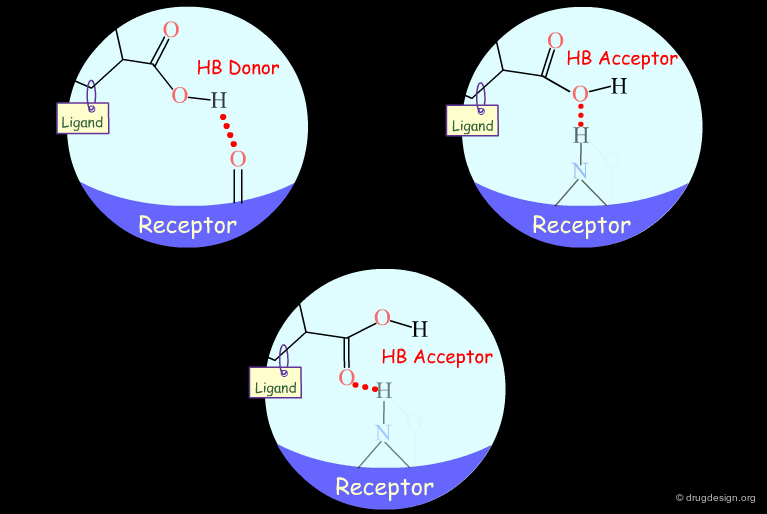
Testing the Existence of H-Bond Interactions¶
It is possible to test for the existence of hydrogen bond interactions with a carboxyl by replacing the COOH group by H, methyl or vinyl.

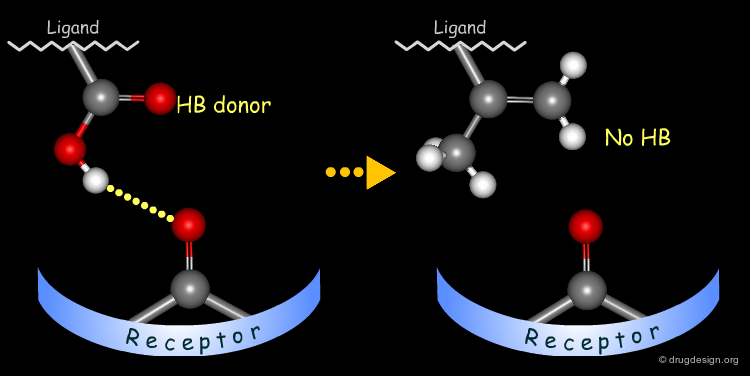
Testing H-Bond Donor Capability of the COOH¶
It is possible to test for the hydrogen bond donor capability with a carboxyl by replacing the COOH group by COOCH3, CHO or CO-CH3.



Example 1: Imidazole Carboxylic Acids¶
In this example, the drop in the biological activity indicates that the carboxyl group may be involved in hydrogen bond interactions or salt bridge interactions with the receptor. Other changes including the volume, the conformational and also the physico-chemical properties of the molecules such as the polar surface area, acidity, hydrophobicity, solubility, solvation etc... must also be envisaged.

articles
Non-Peptide Angiotensin II Receptor Antagonists: Chemical Feature Based Pharmacophore Identification Krovat EM and Langer T J. Med. Chem 46 2003
Example 2: Pyrrolopyrimidines¶
The esterification of the carboxylic acid leads to the complete loss of biological activity. The COOH is therefore important and may be involved as a hydrogen bond donor or in a salt bridge interaction. Other changes including the volume, the geometrical and also the physico-chemical properties of the molecules such as the polar surface area, acidity, hydrophobicity, solubility, solvation etc... must also be envisaged.

articles
4-(Phenylamino)pyrrolopyrimidines: Potent and Selective, ATP Site Directed Inhibitors of the EGF-Receptor Protein Tyrosine Kinase Traxler PM, Furet P, Mett H, Buchdunger E, Meyer T and Lydon N J. Med. Chem 39 1996
Example 3: Peptide-Based Structures¶
The carboxyl group of the molecule shown below may be involved as a hydrogen bond donor or in a salt bridge interaction. Other changes including the volume, the geometrical and also the physico-chemical properties of the molecules such as the polar surface area, acidity, hydrophobicity, solubility, solvation etc... must also be envisaged.

book
Weston GS, Grisostomi C, Cheng H, Lange M, Fiorey M, Cullinane ME, Hardy L, Pallai P, Hartsough DS and Tilton RF Rational Approaches to Drug Design Prous Science 2001
Example 4: Aminoquinolones¶
In the example illustrated here, the ester analog is completely inactive. The free carboxyl function is important and may be involved as a hydrogen bond donor or in a salt bridge interaction. Other changes including the volume, the geometrical and also the physico-chemical properties of the molecules such as the polar surface area, acidity, hydrophobicity, solubility, solvation etc... must also be envisaged.

articles
6-Aminoquinolones as New Potential Anti-HIV Agents Cecchetti V, Parolin C, Moro S, Pecere T, Filipponi E, Calistri A, Tabarrini O, Gatto B, Palumbo M, Fravolini A and Palu G J. Med. Chem 43 2000
Ethers¶
Ether groups are capable of acting only as hydrogen bond acceptors.
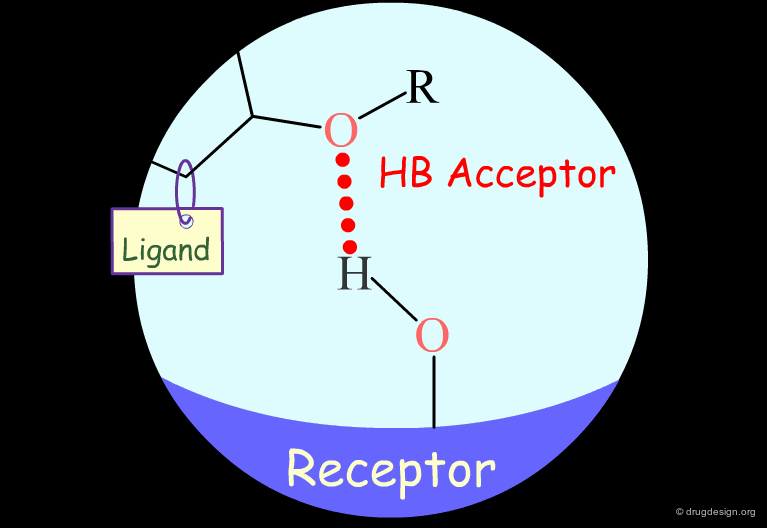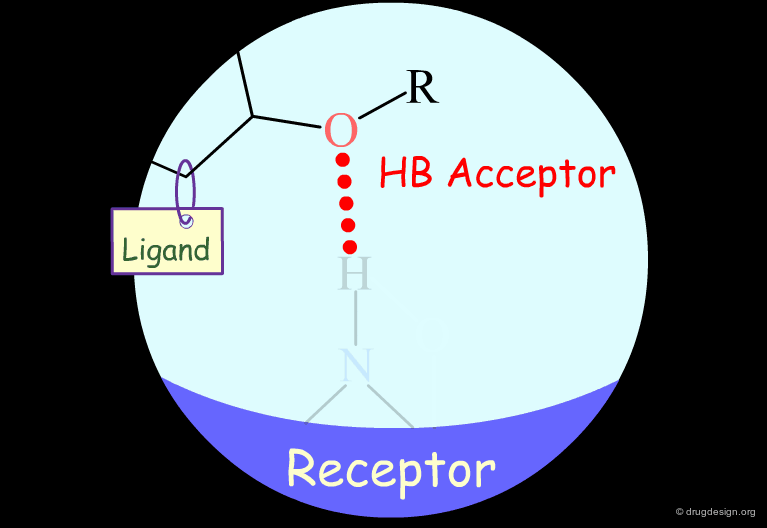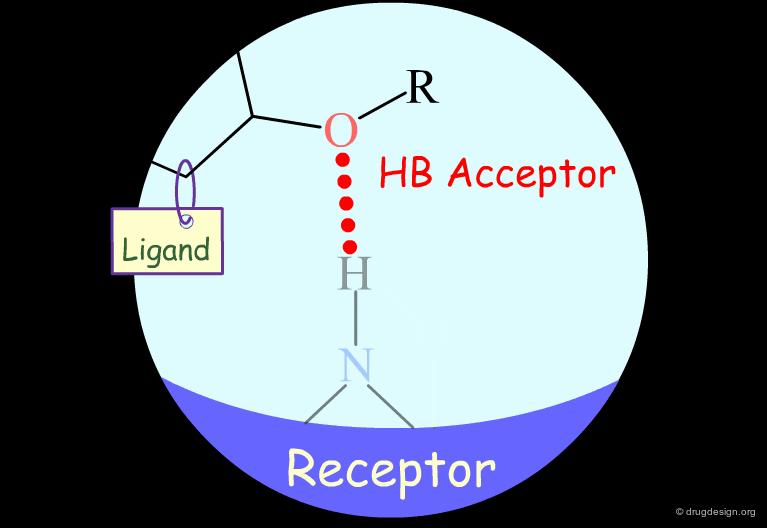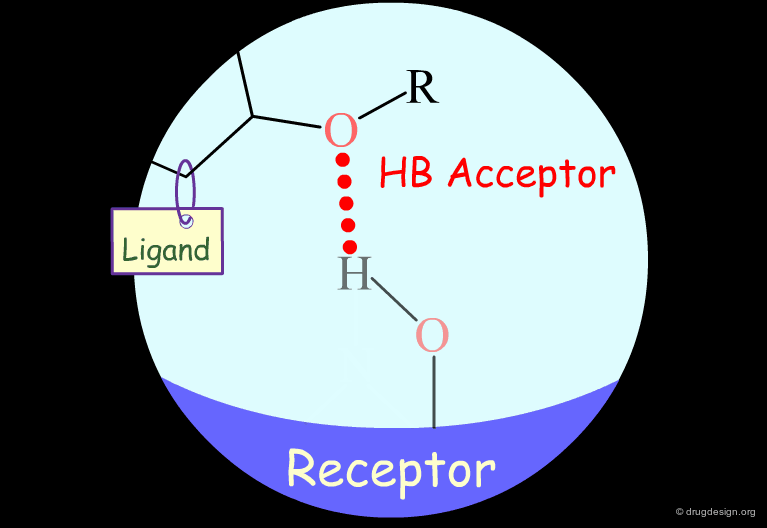
Testing the Existence of H-Bond Interactions¶
To test for the existence of hydrogen bond interactions with an ether fragment, it is possible to make the analog where the oxygen atom is replaced by a carbon.
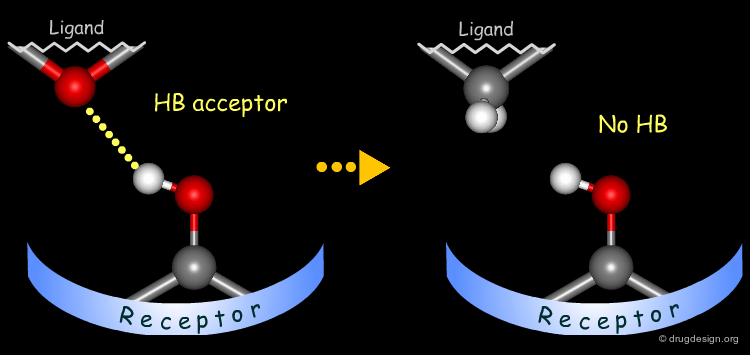
Example 1: Epothilone A¶
In this example (epothilone A), replacing the oxygen in the epoxide moiety with a carbon atom leads to a completely inactive molecule. The oxygen atom may be involved in hydrogen bond interactions with the receptor site. Other changes including the volume and the physico-chemical properties of the molecules such as the polar surface area, hydrophobicity, solubility, solvation etc... must also be envisaged.

articles
Synthesis and biological properties of C12,13-cyclopropyl-epothilone A and related epothilones Nicolaou KC, Finlay MR, Ninkovic S, King NP, He Y, Li T, Sarabia F, Vourloumis D Chemistry and Biology 5 1998
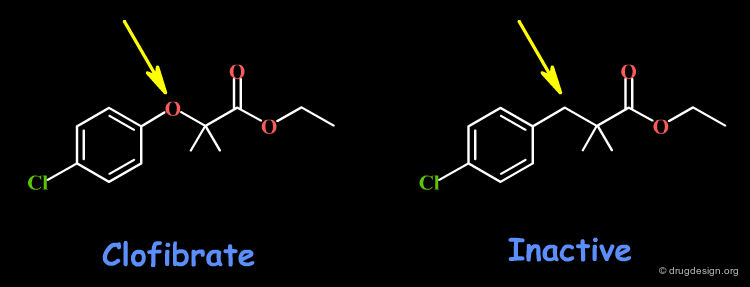
Example 2: Clofibrate¶
Clofibrate is a potent hypolipemic drug, whose activity is lost when replacing the oxygen atom by a carbon: an H-bond interaction might involve this oxygen. Other changes including the volume, the conformational and physico-chemical properties of the molecules such as the polar surface area, hydrophobicity, solubility, solvation etc... must also be envisaged.
book
Witiak DT Clofibrate and Related Analogs: A Comprehensive Review Marcel Dekker 1977
Example 3: Piperidine Renin Inhibitors¶
The optimized molecule shown below (IC50=1.5nM) is active at the nanomolar range, and the addition of two methoxy substituents yields an analog (IC50=0.060nM) with picomolar activities. This is probably due to a better occupancy of a hydrophobic pocket rather than to the formation of additional H-bonds with the receptor (X-Rays).

articles
Renin inhibition by substituted piperidines: a novel paradigm for the inhibition of monomeric aspartic proteinases? Oefner C, Binggeli A, Breu V, Bur D, Clozel JP, D'Arcy A, Dorn A, Fischli W, Gruninger F, Guller R, Hirth G, Marki H, Mathews S, M ller M, Ridley RG, Stadler H, Vieira E, Wilhelm M, Winkler F and Wostl W Chem. Biol 6 1999
Cyano¶
A cyano group is capable of acting only as a hydrogen bond acceptor.

Testing the Existence of H-Bond Interactions¶
To test for the existence of hydrogen bond interactions with a cyano group, it is possible to prepare the analog by replacing the nitrogen atom by a CH (triple bond).

Example 1: Anilinopyrimidines¶
The observed drop in the biological activity indicates that the cyano group may be involved in the formation of a hydrogen bond with the receptor. Other changes including the volume and the physico-chemical properties of the molecules such as the polar surface area, hydrophobicity, solubility, solvation etc... must also be envisaged.

articles
Cyclin-Dependent Kinase 4 Inhibitors as a Treatment for Cancer. Part 1: Identification and Optimisation of Substituted 4,6-Bis Anilino Pyrimidines Beattie JF, Breault GA, Ellston RPA, Green S, Jewsbury PJ, Midgley CJ, Naven RT, Minshull CA, Pauptit RA, Tucker JA and Pease JE Bioorganic and Medicinal Chemistry Letters 13 2003
Example 2: Quinolinecarbonitriles¶
The observed drop in the biological activity indicates that the cyano group may be involved in the formation of a hydrogen bond with the receptor. This hypothesis has been confirmed by molecular modeling and X-ray crystallographic studies.

articles
4-Anilino-6,7-dialkoxyquinoline-3-carbonitrile Inhibitors of Epidermal Growth Factor Receptor Kinase and Their Bioisosteric Relationship to the 4-Anilino-6,7-dialkoxyquinazoline Inhibitors Wissner A, Berger DM, Boschelli DH, Floyd MB, Jr., Greenberger LM, Gruber BC, Johnson BD, Mamuya N, Nilakantan R, Reich MF, Shen R, Tsou HR, Upeslacis E, Wang YF, Wu B, Ye F and Zhang N J. Med. Chem 45 2000
Example 3: Pyrrolopyrimidines¶
The cyano group of the original compound seems to be involved in the formation of a hydrogen bond with the receptor. Other changes including the volume and the physico-chemical properties of the molecules such as the polar surface area, hydrophobicity, solubility, solvation etc... must also be envisaged.

articles
Pyrrolo[2,3-d]pyrimidines Containing an Extended 5-Substituent as Potent and Selective Inhibitors of lck II Burchat AF, Calderwood DJ, Hirst GC, Holman NJ, Johnston DN, Munschauer R, Rafferty P and Tometzkib GB Bioorganic and Medicinal Chemistry Letters 10 2000
Probing Ionic Interactions¶
Principle for Probing Ionic Interactions¶
The method for probing the existence of ionic interactions with a specific functional group consists of preparing analogs where the functional group concerned is unable to make ionic interactions.

Carboxylates¶
For a molecule containing a carboxyl group and when the pH is much greater than the pKa of the corresponding acid, the carboxyl function exists primarily in its ionized, conjugate base form and can therefore establish ionic interactions with positively charged groups. Unless the structure of the complex with the receptor is known, it is not easy to prove the existence of salt-bridge interactions, either with analogs, or by measuring the biological activities at different pHs.
Amines¶
In aqueous solution all amines can exist in an ionic form, however the quaternary ammonium bases are the most strongly ionized. Unless the structure of the complex with the receptor is known, it is not easy to prove the existence of salt-bridge interactions simply by preparing analog compounds that do not have the amine moiety.

Probing Hydrophobic Interactions¶
Importance of Hydrophobic Interactions¶
Hydrophobic interactions represent an important force in the regulation of biological systems. Locally, they correspond to weak and non-directional forces, however their accumulation on entire molecular surfaces may represent forces that can produce substantial effects in terms of affinity and binding.


articles
Computer Graphics in Drug Design: Molecular Modeling of Thyroid Hormone-Prealbumin Interactions Blaney JM, Jorgensen EC, Connolly ML, Ferrin TE, Langridge R, Oatley SJ, Burridge JM and Blake CC J. Med. Chem. 25 1982
Principles for Probing Hydrophobic Interactions¶
The method for probing steric interactions consists of preparing analogs that allow for the introduction of groups having different sizes, in order to determine on the basis of their respective SAR, the shape and the exact nature of the steric interaction concerned.
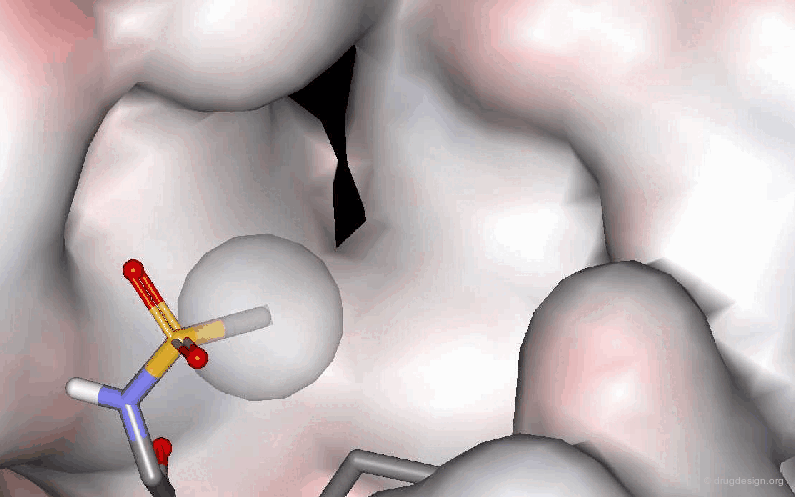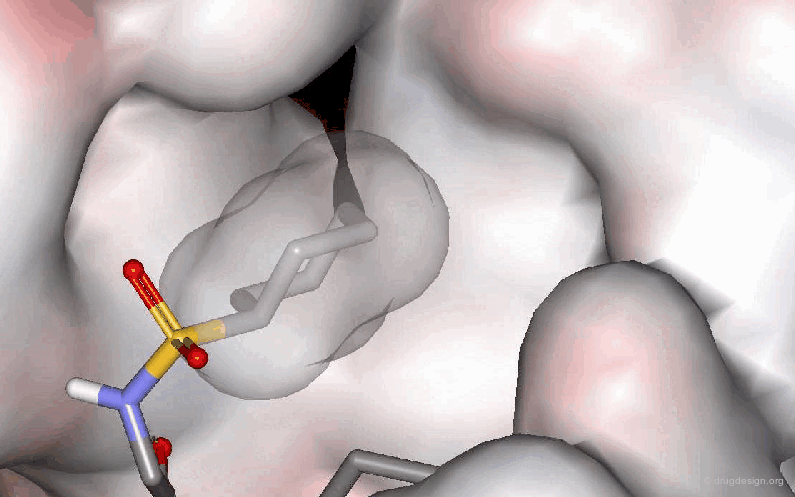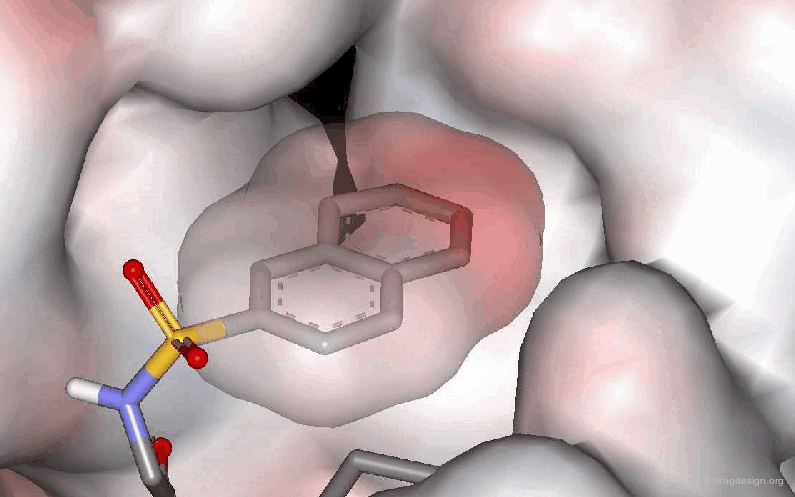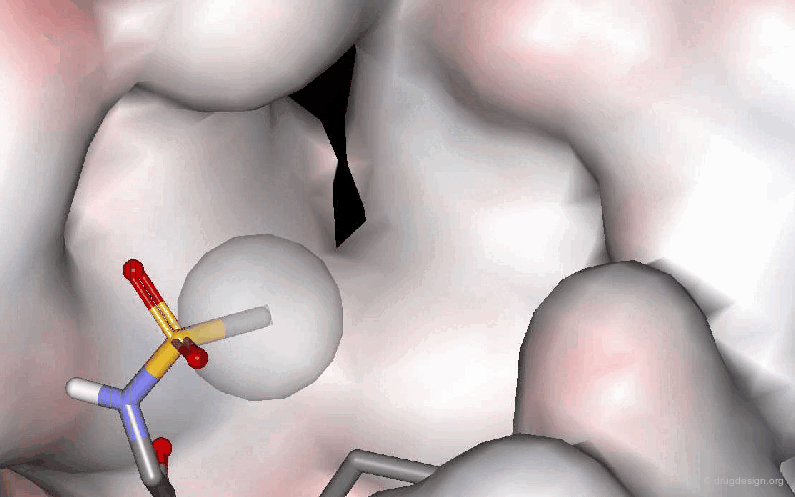
Alteration of Ring Size¶
When an initial ring-containing molecule is suspected of interacting with hydrophobic moieties of the receptor site, a possible way to probe such interactions consists of varying the size of the ring, or fusing additional rings to the original structure.

Example 1: Probing of a Hydrophobic Pocket¶
In the following example, derivatives of the bicyclic benzocyclopentene molecule were prepared to determine the SAR of binding to phospholipase A2. In particular the acenaphthene analog proved to be 400 times more potent than the original compound. Modeling analyses confirm the existence of favorable interactions with the naphthalene moiety deeply buried in a hydrophobic pocket.

articles
Molecular Modeling and Drug Design: Strategies in the Design and Synthesis of Phospholipase A2 Inhibitors Ripka WC, Sipio WJ and Blaney JM Lect. Hetrocycl. Chem 9 1987
Example 2 : Varying Ring Size¶
Various cyclic analogs of the initial isopropyl derivative were prepared and tested. The biological activities proved to increase with the size of the ring. (The higher activity observed for the cyclopentane analog as compared to the cyclohexane can be understood as a better flexibility of the five-membered ring when interacting with the receptor.)

articles
SAR of 4-Hydroxypiperidine and Hydroxyalkyl Substituted Heterocycles as Novel p38 Map Kinase Inhibitors Revesz L, Di Padova FE, Buhl T, Feifel R, Gram H, Hiestand P, Manning U and Zimmerlin AG Bioorganic and Medicinal Chemistry Letters 10 2000
Example 3: Probing a Hydrophobic Pocket¶
The following example describes the SAR exploration of a hydrophobic pocket estimated to be in the vicinity of the amide moiety of the pyridopyrimidinone scaffold. The size of the pocket appears to be limited in length (methyl-cylohexane entirely inactive) and in shape, the most potent compound being the bicyclo-heptane analog (IC50 = 38nM).
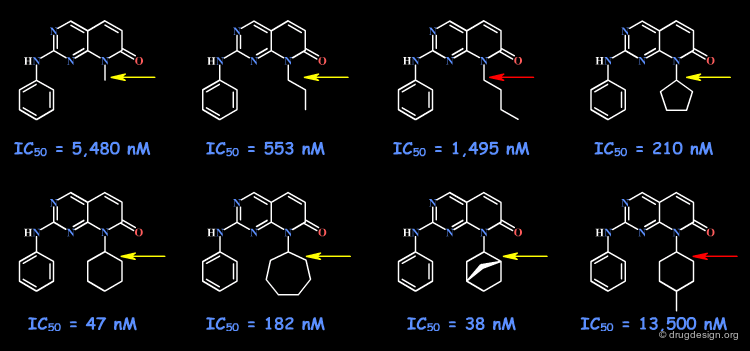
articles
Pyrido[2,3-d]pyrimidin-7-one Inhibitors of Cyclin-Dependent Kinases Barvian M, Boschelli DH, Cossrow J, Dobrusin E, Fattaey A, Fritsch A, Fry D, Harvey P, Keller P, Garrett M, La F, Leopold W, McNamara D, Quin M, Trumpp-Kallmeyer S, Toogood P, Wu Z and Zhang E J. Med. Chem 43 2000
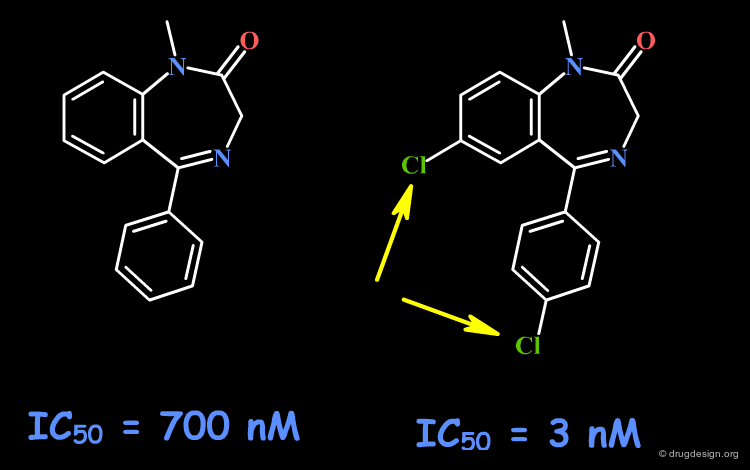
Example 4: Fusion with Additional Rings¶
To probe the existence of hydrophobic interactions with the benzodiazepine receptor, naphthyl-fused diazepine compounds were found to be inactive. Further SAR proved that substantial increased affinities are obtained with molecules containing chlorine atoms. This effect that can be understood either in terms of special hydrophobic interactions, or of polar interactions with the chlorine atoms.
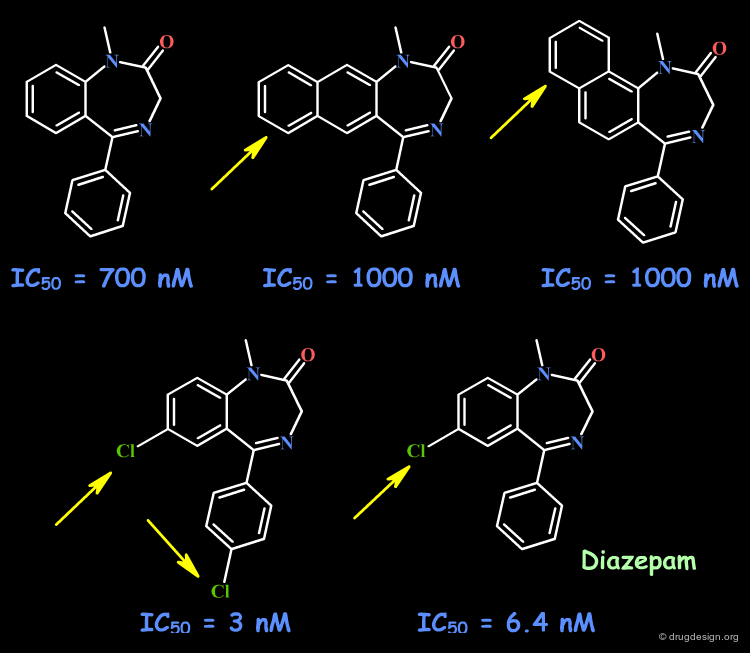
articles
Bioisosterism: A Rational Approach in Drug Design Patani GA and LaVoie EJ Chem. Rev 96 1996
Naturally occurring 2'-hydroxyl-substituted flavonoids as high-affinity benzodiazepine site ligands Huen MSY, Hui KM, Leung JWC, Sigel E, Baur R, Wong JTF and Xue H Biochemical Pharmacology 66 2003
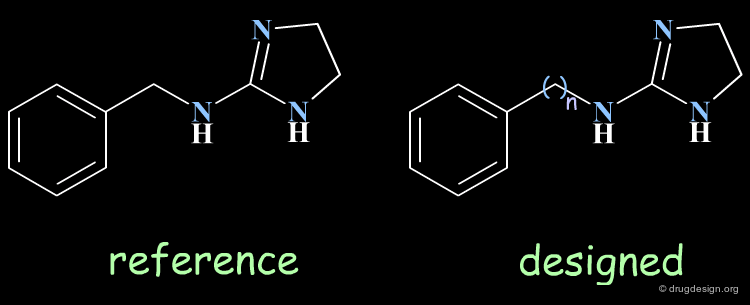
Homologation of Alkyl Chains¶
The increase in the number of carbon atoms in a chain serves to progressively explore the existence and the depth of a pocket located in the region explored.
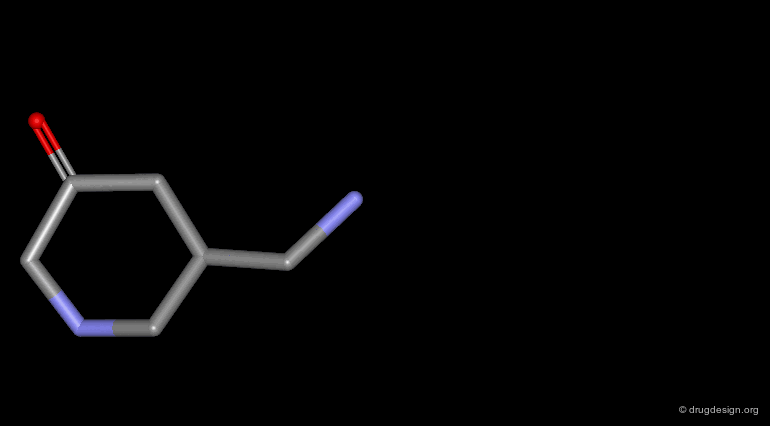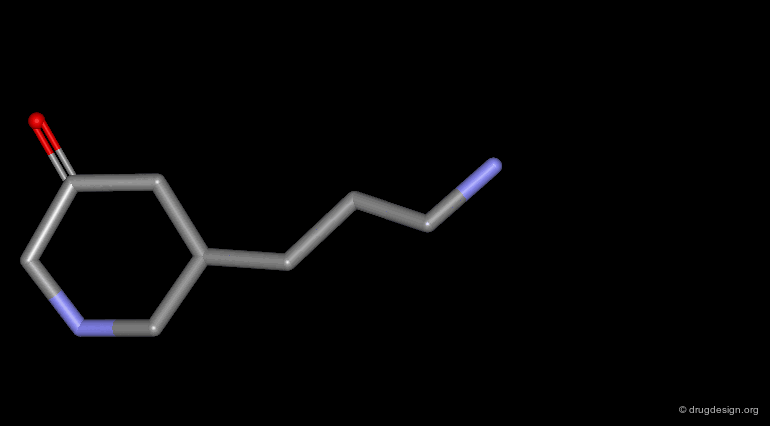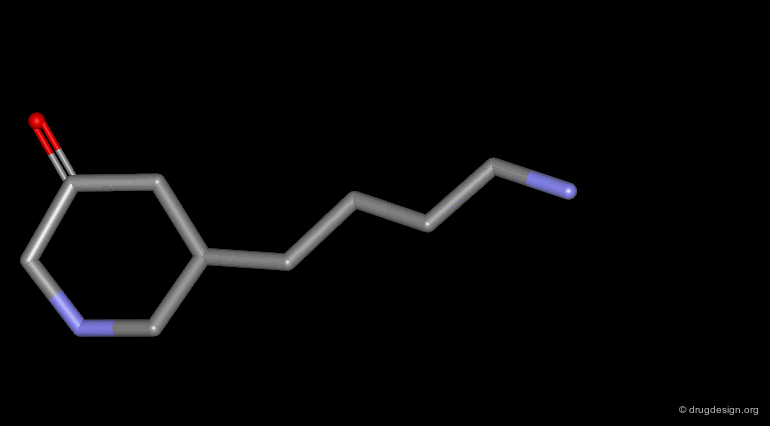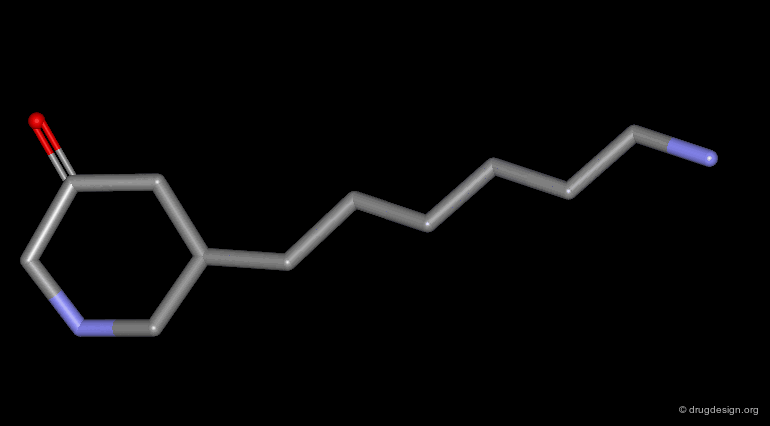
Example 1 of Homologation¶
The hydrogen atom connected to the nitrogen of apomorphine was replaced by methyl, ethyl, n-propyl and n-butyl groups. The best activity was found for the n-propyl analog, whereas the n-butyl demonstrated a great loss in potency.

Example 2 of Homologation¶
In this example the length of the chain was explored, revealing the existence of an optimum for an n-butyl chain, which was further refined with the preparation of branched analogs.
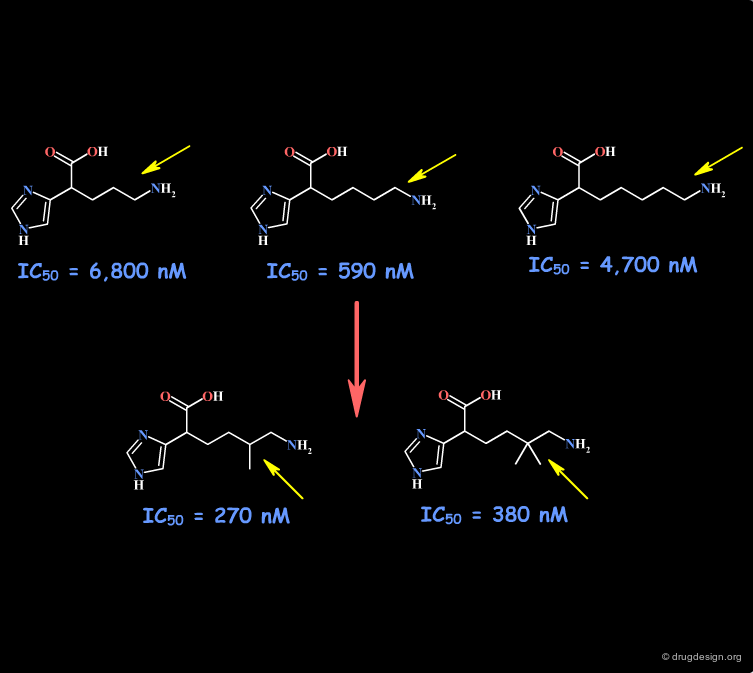
articles
Imidazole acetic acid TAFIa inhibitors: SAR studies centered around the basic P'1 group Nantermet GP, Barrow JC, Lindsley SR, Young M, Mao SS, Carroll S, Bailey C, Bosserman M, Colussi D, McMasters DR, Vacca JP and Selnick HG Bioorganic and Medicinal Chemistry Letters 14 2004
Example 3 of Homologation¶
This is an example of systematic homologation of an initial reference molecule. The SAR reveal the existence of an optimal activity for the n-heptyl chain. The inactivity of the cyclic derivatives indicates that these rings are too bulky to enter into the hypothetical hydrophobic pocket envisaged.

articles
Covalent Modification of Cyclooxygenase-2 (COX-2) by 2-Acetoxyphenyl Alkyl Sulfides, a New Class of Selective COX-2 Inactivators Kalgutkar AS, Kozak KR, Crews BC, Hochgesang GP Jr. and Marnett LJ J. Med. Chem 41 1998
Exploring the Width of a Hydrophobic Pocket¶
It is relatively easy to explore the width of a hydrophobic pocket by making a series of analogs of different sizes.

Example 1: Pyrazoles¶
In this example the steric requirements of a hydrophobic pocket located in the vicinity of the pyrazole heterocycle were tested. The inactivity of the cyclohexyl derivative and the good activity observed for the naphthyl analog suggest that the hydrophobic pocket is flat and narrow.

articles
Pyrazole urea-based inhibitors of p38 MAP kinase: from lead compound to clinical candidate Regan J, Breitfelder S, Cirillo P, Gilmore T, Graham AG, Hickey E, Klaus B, Madwed J, Moriak M, Moss N, Pargellis C, Pav S, Proto A, Swinamer A, Tong L and Torcellini C J. Med. Chem 45 2002
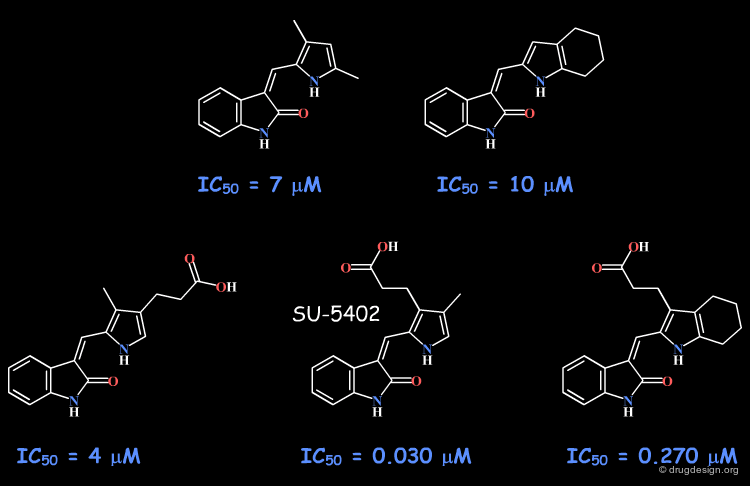
Example 2: Indolinones¶
The indolinone scaffold was discovered by random screening, and this prompted a synthetic program to explore the potential of this scaffold (for protein kinase inhibition). The initial phenyl substituent was replaced by various heterocycles that were all inactive, except the pyrrole analog that showed submicromolar activities.

articles
Synthesis and biological evaluations of 3-substituted indolin-2-ones: a novel class of tyrosine kinase inhibitors that exhibit selectivity toward particular receptor tyrosine kinases Sun L, Tran N, Tang F, App H, Hirth P, McMahon G and Tang C J. Med. Chem 41 1998
Design, synthesis, and evaluations of substituted 3-[(3- or 4-carboxyethylpyrrol-2-yl)methylidenyl]indolin-2-ones as inhibitors of VEGF, FGF, and PDGF receptor tyrosine kinases Sun L, Tran N, Liang C, Tang F, Rice A, Schreck R, Waltz K, Shawver LK, McMahon G and Tang C J. Med. Chem 42 1999
Identification of substituted 3-[(4,5,6, 7-tetrahydro-1H-indol-2-yl)methylene]-1,3-dihydroindol-2-ones as growth factor receptor inhibitors for VEGF-R2 (Flk-1/KDR), FGF-R1, and PDGF-Rbeta tyrosine kinases Sun L, Tran N, Liang C, Hubbard S, Tang F, Lipson K, Schreck R, Zhou Y, McMahon G and Tang C J. Med. Chem 43 2000
Structures of the tyrosine kinase domain of fibroblast growth factor receptor in complex with inhibitors Mohammadi M, McMahon G, Sun L, Tang C, Hirth P, Yeh BK, Hubbard SR and Schlessinger J Science 276(5314) 1997
Variations with the Pyrrole Moiety¶
The scaffold was further explored with a pyrrole fragment. The variations included a tetrahydroindole and a propionic acid functionality at different positions and SU-5402 emerged as a lead candidate for development. The binding of this compound as revealed by X-ray crystallography, is presented in the next page.
X-Ray Data of Indolinone Analog¶
The X-ray data reveal that the indolinone moiety is located in the adenine binding site. The pyrrole and the indolinone rings are nearly coplanar due to the intramolecular H-bond between them. The X-ray data provide evidence on the existence of a flat region that can be reached only with planar moieties.

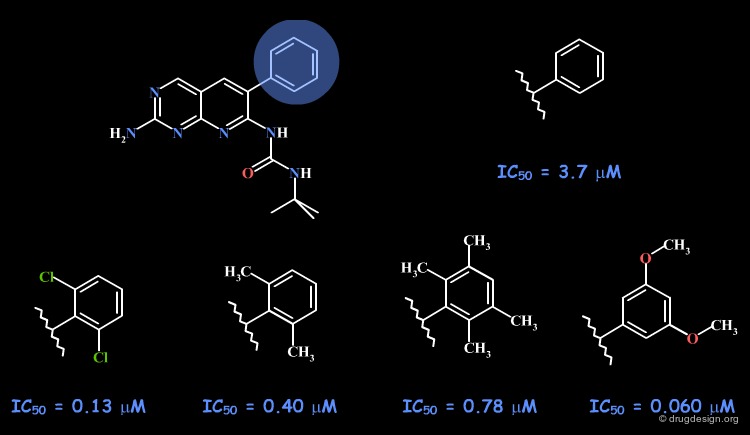
Example 3: Pyridopyrimidines¶
With an initial hit having an IC50 of 3.7 µM, the SAR variations indicated the existence of a flat pocket occupied by the free phenyl substituent. It was difficult to have nanomolar activities, however the 3-5-dimethoxy analog showed an IC50 of 60 nM. The orientation of this compound in the active site, as revealed by X-ray crystallography is presented in the next page.
articles
Structure-activity relationships for a novel series of pyrido[2,3-d]pyrimidine tyrosine kinase inhibitors Hamby JM, Connolly CJ, Schroeder MC, Winters RT, Showalter HD, Panek RL, Major TC, Olsewski B, Ryan MJ, Dahring T, Lu GH, Keiser J, Amar A, Shen C, Kraker AJ, Slintak V, Nelson JM, Fry DW, Bradford L, Hallak H and Doherty AM J. Med. Chem 40 1997
Crystal structure of an angiogenesis inhibitor bound to the FGF receptor tyrosine kinase domain Mohammadi M, Froum S, Hamby JM, Schroeder MC, Panek RL, Lu GH, Eliseenkova AV, Green D, Schlessinger J and Hubbard SR EMBO J 17 1998
X-Ray Data with Pyridopyrimidine Structure¶
The X-ray data provide evidence on the existence of a flat region that is difficult to reach, being almost perpendicular to the pyrido-pyrimidine scaffold. This rather big and flat pocket, can be filled with fragments such as the 3-5-dimethoxy analog.

Probing the Polarity of a Pocket¶
Dealing with a hydrophobic pocket does not mean that this pocket is 100% hydrophobic. It is possible to find heteroatoms belonging to the backbone or to side-chains of the receptor site. SAR are used to probe their existence and their location.

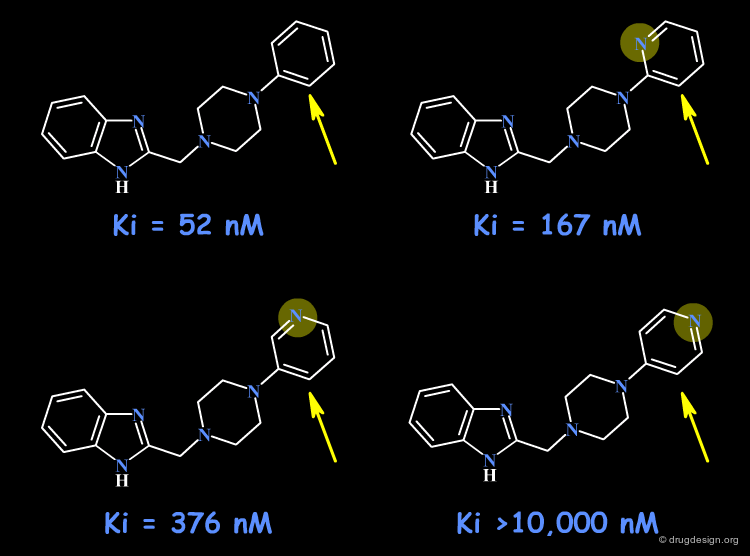
Example: Dopamine Antagonists¶
In the following example a hydrophobic pocket has been found in the prolongation of the piperazine ring. Following the satisfactory results obtained for the phenyl analog (IC50 = 52 nm) the three pyridyl isomers were prepared and proved to be less active. It is interesting to note the complete inactivity of the 4-pyridyl analog, which may reveal the presence of unfavorable polar interactions (e.g. with a C=O) with the nitrogen atom.
articles
Dopamine D4 ligands and models of receptor activation: 2-(4-pyridin-2-ylpiperazin-1-ylmethyl)-1H-benzimidazole and related heteroarylmethylarylpiperazines exhibit a substituent effect responsible for additional efficacy tuning Stewart AO, Cowart MD, Moreland RB, Latshaw SP, Matulenko MA, Bhatia PA, Wang X, Daanen JF, Nelson SL, Terranova MA, Namovic MT, Donnelly-Roberts DL, Miller LN, Nakane M, Sullivan JP and Brioni JD J. Med. Chem 47 2004
Probing Other Interactions¶
Halogens¶
Halogen atoms are able to produce various sorts of interactions. Directly, they may be implicated in hydrophobic or polar interactions; and indirectly they are able to change the polarity of contiguous atoms or conjugated systems (e.g. atoms of aromatic rings). Halogen interactions are observed with many groups (amino, cyano, nitro, imino and other halogen atoms) and also with π systems such as Phe, His, Trp, Tyr.
articles
A novel pyrazoloquinoline that interacts with brain benzodiazepine receptors: characterization of some in vitro and in vivo properties of CGS 9896 Gee KW, Yamamura HI Life Sci 30 1982
Example 1: COX-2¶
In this example the substitution by a chlorine atom leads to a compound which is by far more potent than the unsubstituted derivative. This can be interpreted by the existence of an empty space corresponding to the volume of a chlorine atom. The introduction of a chlorine atom can increase the binding affinity by establishing additional van der Waals interactions with the protein. Other reasons may be due to changes in the volume, the conformation or in the physico-chemical properties of the molecule.

articles
Estimation of binding affinities for celecoxib analogues with COX-2 via Monte Carlo-extended linear response Wesolowski SS and Jorgensen WL Bioorg. Med. Chem. Lett 12 2002
Example 2: MDM2¶
Modeling analyses indicated the existence of an empty space between the peptide and the protein (Mdm2) corresponding to the volume of a methyl or a chlorine atom. Substituting the hydrogen by a methyl or a chlorine led to very potent compounds, whose activities were predicted (before being synthesized) by the formation of favorable steric interactions with the protein.

articles
Discovery of potent antagonists of the interaction between human double minute 2 and tumor suppressor p53 Garcia-Echeverria C, Chene P, Blommers MJ and Furet P J. Med. Chem 43 2000
Example 3: Glycine Antagonists¶
Kynurenic acid is a poor glycine antagonist (IC50 = 41 µM), however, this molecule proved to be an excellent initial lead. The introduction of a chlorine in position 7 resulted in a compound 70-fold more potent. The addition of a iodine in position 5 led to a compound which is 1000-fold more potent than kynurenic acid itself (IC50 = 32 nM). This can be interpreted in terms of favorable electronic effects of the halogen atoms and good interactions with the receptor site.
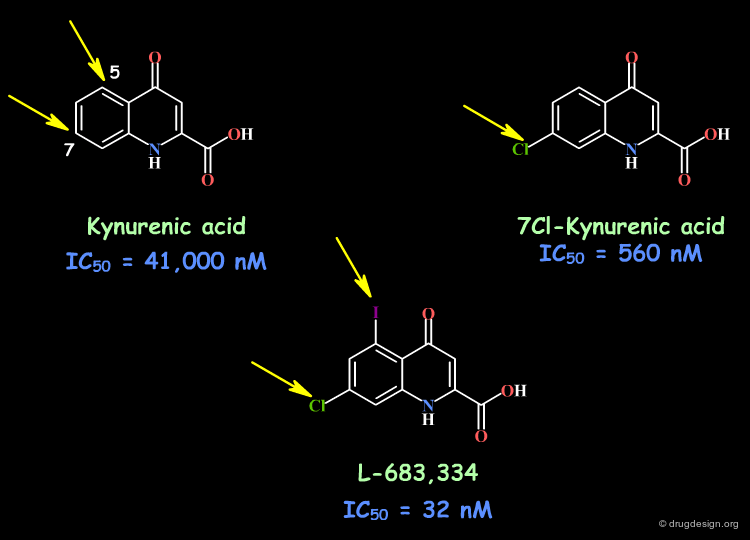
articles
7-Chlorokynurenic acid is a selective antagonist at the glycine modulatory site of the N-methyl-D-aspartate receptor complex Kemp JA, Foster AC, Leeson PD, Priestley T, Tridgett R, Iversen LL and Woodruff GN Proc. Natl. Acad. Sci. U.S.A 85 1988
book
Lesson PD, Carling RW, Kulagowski JJ, Mawer IM, Moore KW, Moseley AM, Rowley M, Smith JD, Stevenson GI, Williams BJ, Baker R, Foster AC, Kemp JA and Tricklebank MD Prespectives in Medicinal Chemistry Verlag Helvetica Chimica Acra, Basel 1993
Example 4: EGF-R Kinase Inhibitors¶
An important milestone in the search for EGF-R kinase inhibitors was the discovery of the 4-anilinoquinazoline pharmacophore. SAR showed that very potent compounds are obtained when substituting the anilino in meta, with halogen atoms (also with an ethynyl). It was later proved (initially by modeling and later by X-rays) that the substituted inhibitor creates favorable steric interactions with a hydrophobic chimney of the EGF-R-ATP binding domain of the kinase.

articles
The 4-anilinoquinazoline class of inhibitors of the erbB family of receptor tyrosine kinases Denny WA Il Farmaco 56 2001
Tyrosine kinase inhibitors. 16. 6,5,6-tricyclic benzothieno[3, 2-d]pyrimidines and pyrimido[5,4-b-] and -[4,5-b]indoles as potent inhibitors of the epidermal growth factor receptor tyrosine kinase Showalter HD, Bridges AJ, Zhou H, Sercel AD, McMichael A, Fry DW J. Med. Chem 42 1999
Chemical Inhibitors of Protein Kinases Bridges AJ Chem. Rev 101 2001
Probing Aromatic Ring Positions¶
When dealing with aromatic rings, one of the most popular ways in medicinal chemistry for deriving SAR is to probe the aromatic ring positions.

KDR Inhibitors¶
In this series, the ortho, meta and para substitutions were probed with a methoxy group. The SAR yielded the following order of preference: meta>ortho>para. An additional methoxy group was further introduced in the second meta position, and led to an inhibitor which proved to be even more potent.

articles
2,4-disubstituted pyrimidines: a novel class of KDR kinase inhibitors Manley PJ, Balitza AE, Bilodeau MT, Coll KE, Hartman GD, McFall RC, Rickert KW, Rodman LD and Thomas KA Bioorg. Med. Chem. Lett 13 2003
Anthranilamide - Factor Xa Inhibitors¶
Modifications of the three aromatic groups of compound 1 (discovered by HTS) were investigated. Starting by varying the substituents in the aniline ring, the following SAR were observed (subnanomolar activities for the 4-Br and 4-Cl analogs).

articles
Structure-activity relationships of substituted benzothiophene-anthranilamide factor Xa inhibitors Chou YL, Davey DD, Eagen KA, Griedel BD, Karanjawala R, Phillips GB, Sacchi KL, Shaw KJ, Wu SC, Lentz D, Liang AM, Trinh L, Morrissey MM and Kochanny MJ Bioorg. Med. Chem. Lett 13 2003
Exploration of Anthranilamide Substitution¶
Keeping a 4-Cl substituent, the substitution of the central anthranilamide ring was then explored (with mono and di-substitutions). Subnanomolar activities are observed, only for a methyl or a chlorine at the C-5 position.

Replacement of the Benzothiophene Ring¶
Finally, the substitution or the replacement of the benzothiophene ring were explored, but did not reveal new lead structures.

Useful Data Collected for Understanding SAR¶
Systematic variations were made on the three aromatic groups of the initial compound. In the original work, the SAR were analyzed in terms of the properties of the substituents (e.g. size, electron-withdrawing, electron-donating, H-bond donating, isosteric, polar protic etc...). Modeling and crystallographic studies should complement the knowledge so far derived, and contribute to a better understanding of the SAR.

Tetrahydroisoquinoline - Serotonin 5-HT2A Ligands¶
Modifications of the initial tetrahydroisoquinoline structure were envisaged. The substitution at C-7 was first explored, and the 7-methyl analog proved to be the most potent compound of this series.

articles
The influence of substitution at aromatic part of 1,2,3,4-tetrahydroisoquinoline on in vitro and in vivo 5-HT(1A)/5-HT(2A) receptor activities of its 1-adamantoyloaminoalkyl derivatives Bojarski AJ, Mokrosz MJ, Minol SC, Koziol A, Wesolowska A, Tatarczynska E, Klodzinska A and Chojnacka-Wojcik E Bioorg. Med. Chem. Lett 12 2002
Preferred Position for the Chlorine¶
Varying the position of the chlorine atom indicated a preference for the substitution at C-5.
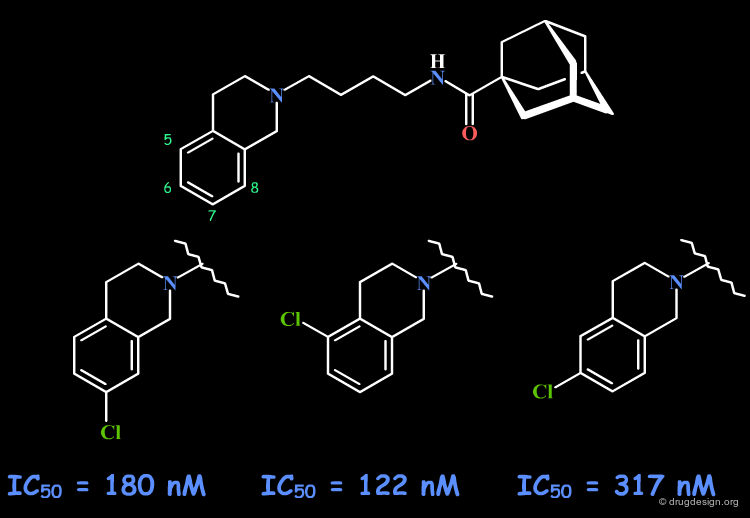
Most Potent Analogs¶
Additional mono and di-substituted analogs were also prepared. The most potent compound proved to be the 5-Methoxy, 8-Br analog (IC50 = 40 nM).

Modifications to Alter the Geometry of the Ligand¶
Modification to Alter the Geometry of the Ligand¶
The method for probing the 3D stereochemical requirements of a receptor site consists of preparing analogs that allow for the introduction of groups of different sizes, shape, chirality, polarity and physico-chemical properties, in order to determine the exact nature of the 3D requirements for interacting with the receptor, in a more general perspective than with elementary SAR analyses.

The Amide Function¶
Acyclic secondary amide moieties are always found in the trans (Z) geometry. However, tertiary amides exist as a mixture of E and Z isomers: this is an important property of acyclic tertiary amides, which should not be overlooked when exploring SAR. Moreover, when the amide group is located in the center of the molecule, this situation can be exploited to explore the requirements of two entirely different regions of the receptor.
![]()
SAR Example with an Amide Moiety¶
In the following example the tertiary amide analog is 30,000 times more potent than the secondary amide. Based on NMR analyses of the chemical shift of the proton in position 2 of the quinoline, the SAR were interpreted with the bioactive conformation in out-of-plane orientation of the carbonyl. However, it is also possible to consider the existence of a cis geometry (see bottom view) that is only possible for the tertiary amide, which can reach an entirely new pocket.
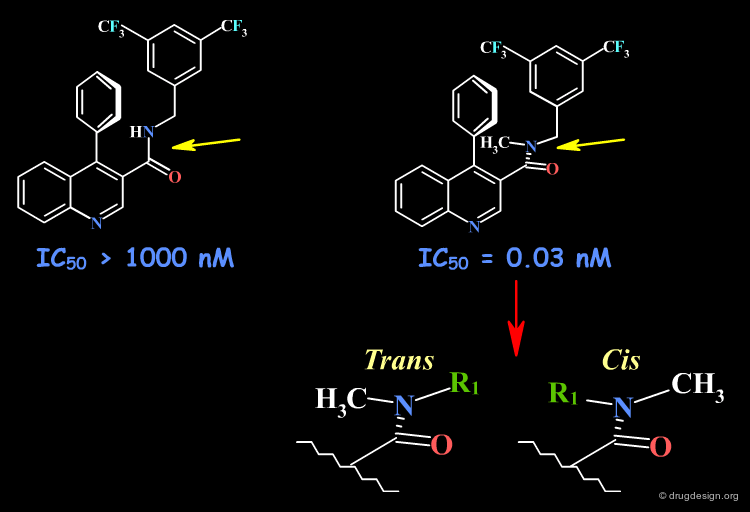
articles
A non-peptide NK1 receptor agonist showing subpicomolar affinity Cappelli A, Giuliani G, Pericot Mohr Gl G, Gallelli A, Anzini M, Vomero S, Cupello A, Scarrone S, Matarrese M, Moresco RM, Fazio F, Finetti F, Morbidelli L and Ziche M J. Med. Chem 47 2004
Ortho Substitution of Aromatic Ring¶
When considering ortho, meta and para substitutions of aromatic rings, ortho analogs introduce conformational changes in the molecule due to steric repulsions (even with small substituents such as CH3 or Cl). In meta and para analogs the conformation remains unchanged.
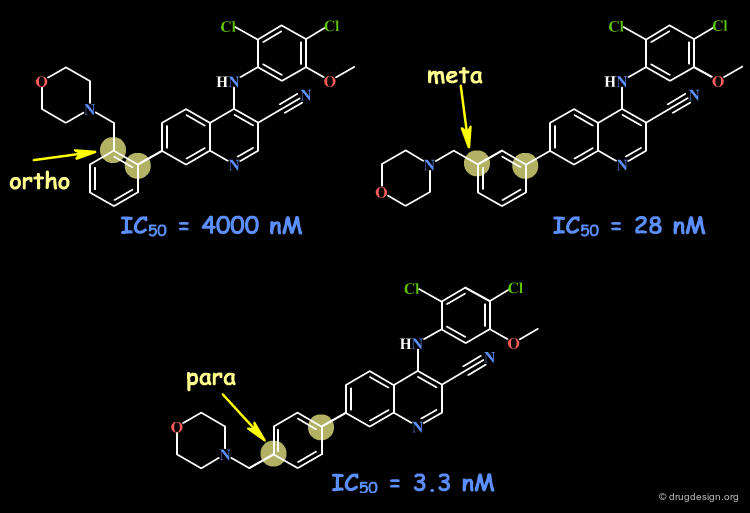
articles
Substituted 4-anilino-7-phenyl-3-quinolinecarbonitriles as Src kinase inhibitors Berger D, Dutia M, Powell D, Wissner A, DeMorin F, Raifeld Y, Weber J and Boschelli F Bioorg. Med. Chem. Lett 12 2002
Cis-Trans Isomers¶
In a molecule containing an acyclic double bond, it might be useful to assess the properties of the E and Z isomers. Although the other isomer is not always easy to prepare, the measurement of its biological activity might be highly informative as to the global stereochemical requirements of the receptor site.
![]()
Example 1: Cis and Trans Isomers¶
The Z stereochemistry was suspected to be the bioactive geometry of methyl-idenyl-indolin-2-one derivatives. To test this hypothesis the 4'-chlorine molecule (right), and the 4'-chlorine 1'-methyl derivative (left), were conceived with geometrical considerations that were confirmed to be correct. N.M.R. studies indicate a Z geometry that is favored by an internal hydrogen bond for the first molecule, and the E geometry for the second compound. The biological tests confirmed that a Z stereochemistry is needed for having active molecules.
![]()
articles
Synthesis and biological evaluations of 3-substituted indolin-2-ones: a novel class of tyrosine kinase inhibitors that exhibit selectivity toward particular receptor tyrosine kinases Sun L, Tran N, Tang F, App H, Hirth P, McMahon G and Tang C J. Med. Chem 41 1998
Z and E Isomers are in Equilibrium !¶
It is worth noticing that for the structures of the previous page, there are no separate Z and E isomers: they are in equilibrium! In this conjugated system, the double bond becomes single in the enolic tautomer form 2, which then allows the rotation about this bond. The isomerization is thermodynamically controlled with an E/Z ratio depending on the energies of the two isomers (for the R group concerned).
![]()
Example 2: Cis and Trans Isomers¶
The bioactive conformation for binding to the GABAc proved to correspond to the trans isomer. In the GABA series the trans (TACA) isomer is more potent than the cis (CACA); the inferior activity of GABA can be understood in terms of the free rotation around the central bond. In the methylphosphinic analogs it is also observed that the trans isomer (CGP44530) is more potent than the cis (CGP70523); however the good biological activity observed for the unsaturated CGP35024 is difficult to interpret.
![]()
articles
Unsaturated phosphinic analogues of gamma-aminobutyric acid as GABA(C) receptor antagonists Chebib M, Vandenberg RJ, Froestl W, Johnston GA Eur. J. Pharmacol 329 1997
Alter Stereochemistry¶
The following is a simple example showing that a simple stereochemical change can lead to molecules with very different biological properties. Cis-platin is an active compound used to treat cancer; trans-platin, although very similar is completely inactive.
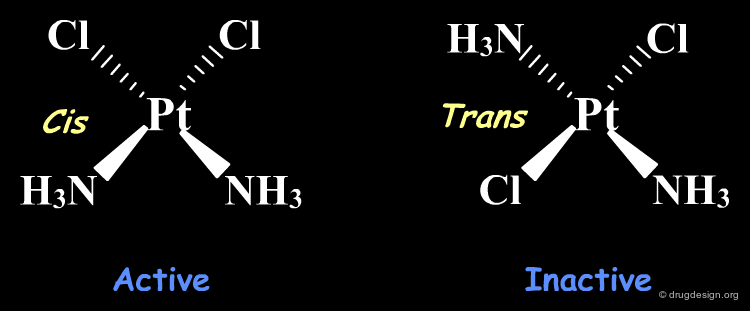
Rigid Analogs: SAR Principle¶
When comparing the SAR between flexible and rigid analogs, the geometrical aspects are of primary importance. Reciprocally, it is always possible to introduce in a structure, an additional rigidity to explore the consequence in the SAR, and derive useful knowledge on the 3D requirements of the active site.

articles
Lowering the Entropic Barrier for Binding Conformationally Flexible Inhibitors to Enzymes Khan AR, Parrish JC, Fraser ME, Smith WW, Bartlett PA, and James MN Biochemistry 37 1998
Energetic and Entropic Factors Determining Binding Affinity in Protein-Ligand Complexes Klebe G, Bohm HJ J. Recept Signal Transduct Res. 17 1997
The Cost of Conformational Order: Entropy Changes in Molecular Associations Searle MS and Williams DH J. Am. Chem.Soc. 14 1992
Example 1: GnRH Antagonists Rigidification¶
Constraining the two methoxy groups in a benzodioxane-1,4 ring results in a molecule with improved affinity and binding. In addition to the geometrical changes, more favorable physico-chemical properties and also desolvation energies should not be excluded.

articles
Synthesis and structure-activity relationships of 1-arylmethyl-5-aryl-6-methyluracils as potent gonadotropin-releasing hormone receptor antagonists Guo Z, Zhu YF, Gross TD, Tucci FC, Gao Y, Moorjani M, Connors PJ Jr, Rowbottom MW, Chen Y, Struthers RS, Xie Q, Saunders J, Reinhart G, Chen TK, Bonneville AL and Chen C J. Med. Chem 47 2004
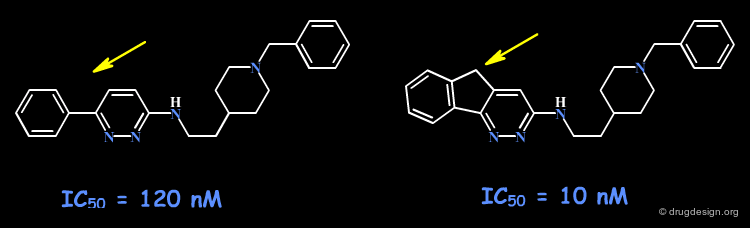
Example 2: AChE Rigidification¶
The rigidification of the free aromatic ring in a tricyclic structure leads to a molecule with increased potency. In addition to the geometrical changes, more favorable physico-chemical properties and also desolvation energies should not be excluded.
book
Wermuth CG Rational Approaches to Drug Design Prous Science 2001
Example 3: p56-lck Inhibitors Rigidification¶
In the example shown below, the rigidification of the central rotatable bond leads to an ethylenic analog which is more potent than the saturated one. This gives useful information on the bioactive conformation required for binding to the receptor.

articles
Non-amine based analogues of lavendustin A as protein-tyrosine kinase inhibitors Smyth MS, Stefanova I, Hartmann F, Horak ID, Osherov N, Levitzki A and Burke TR Jr J. Med. Chem 36 1993
Example 4: Angiotensin-II Receptor Antagonists¶
The n-butyl fragment of the imidazole compound shown below was replaced by the corresponding trans-butene moiety and resulted in a much more potent compound. This indicates that an extended conformation is required for binding to the receptor.

articles
Non-Peptide Angiotensin II Receptor Antagonists: Chemical Feature Based Pharmacophore Identification Krovat EM and Langer T J. Med. Chem 46 2003
Example 5: Dopaminergics Rigidification¶
Dopamine has two trans rotameric forms that can be incorporated in different structurally constrained systems. Different biological activities were observed for the following tetralin rigid analogs. Such rigid scaffolds are excellent leads for further SAR analyses to probe the exact requirements for binding to the different sub-populations of dopamine receptors.

articles
Further Characterization of Structural Requirements for Agonists at the Striatal Dopamine D-1 Receptor. Studies with a Series of Monohydroxyaminotetralins on Dopamine-Sensitive Adenylate Cyclase and a Comparison with Dopamine Receptor Binding Seiler MP and Markstein R Mol Pharmacol. 22 1982
Further Characterization of Structural Requirements for Agonists at the Striatal Dopamine D2 Receptor and a Comparison with those at the Striatal Dopamine D1 receptor. Studies with a Series of Monohydroxyaminotetralins on Acetylcholine Release from Rat Striatum Seiler MP and Markstein R Mol Pharmacol. 26 1984
book
Seiler MP, Bolsterli JJ, Floersheim P, Hagenbach A, Markstein R. Pfaffli P, Widmer A, and Wuthrich H Perspectives in Medicinal Chemistry VCH 1993
Examples 6: Pseudo Rings¶
It is possible to mimic the presence of a ring by designing a pseudo-cycle, whose "closure" will be an intramolecular hydrogen bond in the structure concerned.

articles
Conformationally restricted analogues of remoxipride as potential antipsychotic agents Norman MH, Kelley JL and Hollingsworth EB J. Med. Chem 36 1993
Anthranilamides¶
The following illustrates how an anthranilamide scaffold was successfully designed to resemble to an initial anilinophtalazine compound. The intramolecular H-bond between the aniline-NH and the benzanilide C=O favors the conformation which mimics well the reference bicyclic structure (see 3D view).


articles
Identification of a new chemical class of potent angiogenesis inhibitors based on conformational considerations and database searching Furet P, Bold G, Hofmann F, Manley P, Meyer T, Altmann KH Bioorg. Med. Chem. Lett 13 2003
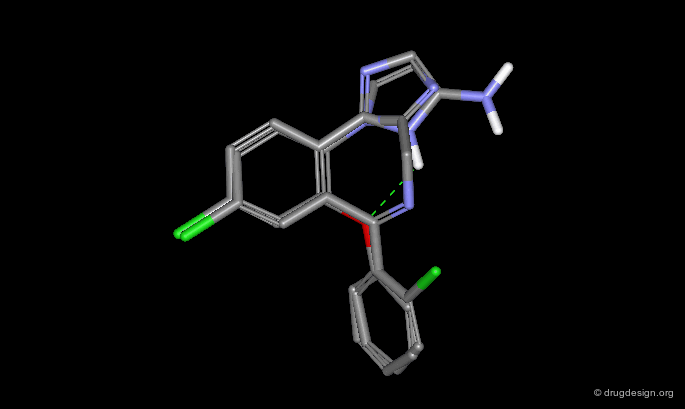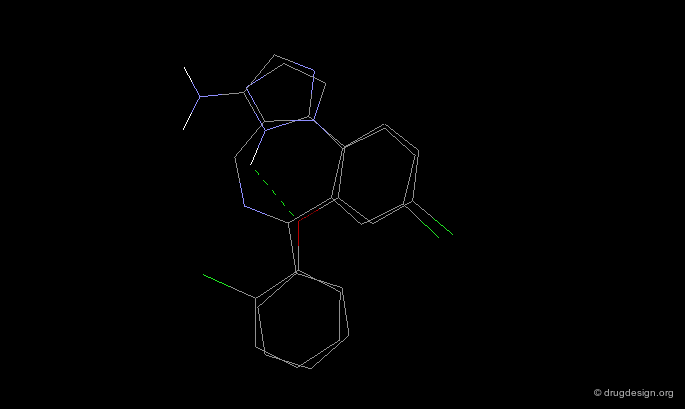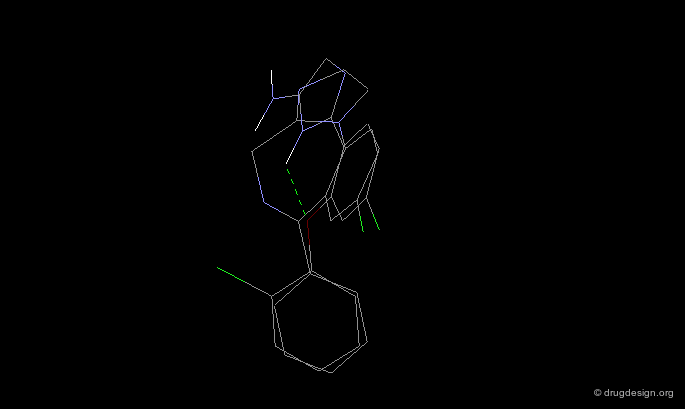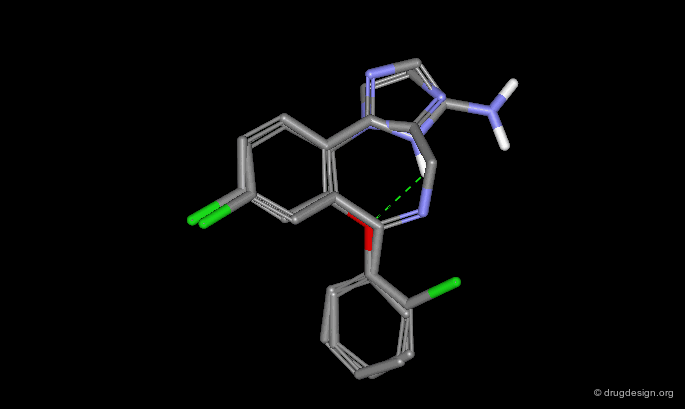
Phenoxyphenyltriazoles¶
The phenyl-triazole derivative shown below was conceived to mimic the structure of benzodiazepine receptor antagonists such as estazolam. Its seven-membered diazepine ring is mimicked by a virtual ring, as can be seen from both the 2D and 3D superpositions.

articles
Design and Synthesis of 4H-3-(2-Phenoxy)phenyl-1,2,4-triazole Derivatives as Benzodiazepine Receptor Agonists Akbarzadeh T, Tabatabai SA, Khoshnoud MJ, Shafaghic B and Shafiee A Bioorganic and Medicinal Chemistry 11 2003
Salicylanilides¶
The salicylanilide scaffold shown here was designed to match a reference quinazoline structure (assuming the formation of the proper H-bond, see 3D view). The "morphing" operation allowed for the discovery of a new type of scaffold, to be further optimized.

articles
Salicylanilides as inhibitors of the protein tyrosine kinase epidermal growth factor receptor Liechti C, Sequin U, Bold G, Furet P, Meyer T, Traxler P Eur. J. Med. Chem 39 2004
Tyrosine kinase inhibitors. 8. An unusually steep structure-activity relationship for analogues of 4-(3-bromoanilino)-6,7-dimethoxyquinazoline (PD 153035), a potent inhibitor of the epidermal growth factor receptor Bridges AJ, Zhou H, Cody DR, Rewcastle GW, McMichael A, Showalter HD, Fry DW, Kraker AJ, Denny WA j. Med. Chem 39 1996
A diazine heterocycle replaces a six-membered hydrogen-bonded array in the active site of scytalone dehydratase Hodge CN and Pierce J Bioorganic and Medicinal Chemistry Letters 3 1993
Example 7: Good and Bad Rigidification¶
The rigidification of the two psychedelic drugs, DOB and mescaline are presented here. The rigidification of the methoxy groups in DOB leads to more active compounds, whereas the similar idea in mescaline yields inactive molecules. However, the biological activities are based on animal models, and the interpretations are tricky.
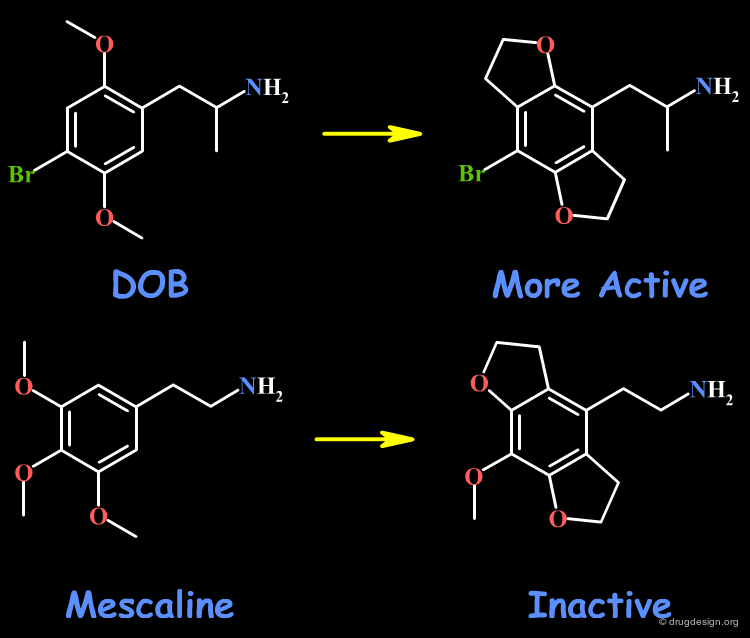
articles
The Medicinal Chemistry of Phenethylamine Psychedelics Nichols DE The Heffter Review of Psychedelic Research 1 1998
Example 8: Rigidification of a Flexible Molecule¶
The following example illustrates a successful attempt to reveal the probable bioactive conformation of a flexible molecule. The introduction of an additional ring (2 torsion angles blocked) results in favorable geometry of the molecule, as indicated by the increase in potency of the tricyclic compound. Note that due to steric repulsions, the poorly active ortho methyl analog cannot adopt the conformation of the tricyclic structure.
articles
Similarity and dissimilarity: a medicinal chemist's view Kubinyi H Perspectives in Drug Discovery and Design 9-11 1998
Flexible Analogs: SAR Principle¶
It is possible to introduce additional flexibility to a cyclic structure by suppressing one ring. This approach is generally used when the initial lead is not attractive enough; the main motivation is to find another starting point.
Ring Suppression: Doxepin¶
The structure of doxepin has been used as a starting point for the design of non-polycyclic analogs. Cutting one of the bonds in this structure results in an analog that has one ring less and with the same type of biological activities and can serve as a starting point for deriving new SAR.

Alteration of Interatomic Distances¶
The systematic alteration of interatomic distances between pharmacophore elements allows us to both explore the requirements of a receptor site and to optimize a series of molecules. The following is a typical analog program conducted for the systematic exploration of the biological properties of a reference active compound to probe the 3D requirements of the receptor site.
Alteration of the Stereochemistry of the Ligand¶
Altering the stereochemistry of an active compound is far from being the priority in SAR. When enantiomers have different activities, or if the synthesis of a natural compound (e.g. alkaloid, steroid etc...) allows it, analogs with inverted centers may be considered. For example retroprogesterone is a synthetic steroid which is more active than natural progesterone.

articles
Enantiomeric Steroids: Synthesis, Physical and Biological Properties Biellmann JF Chem. Rev 103 2003
Title Westerhof P Rec. Trav. Chim 79 1960
A new class of hormonally active steroids Rwwrnik EH, Scholer HF, Westerhof P, Querido A, Kassenaar AA, Diczfalusy E, Tillinger KC Naturte 186 1960
The x-ray structure analysis of 17-beta-bromoacetoxy-9-beta, 10-alpha-androst-4-en-3-on Oberhansli WE, Robertson JM Helv. Chim. Acta 50 1967
book
Miyake T and Rooks WH II Methods in Hormone Research Vol V, Part C Academic Press NY 1966
Complexity of SAR Analyses¶
Complexity of the Structures Concerned¶
Sometimes an elementary change (the replacement of an oxygen atom by a carbon, in the example below) cannot be interpreted easily. It may be due to a combination of many effects, such as for example the loss of an important intermolecular interaction, and/or the absence of an internal H-bond which favors the bioactive conformation, and/or changes in desolvation, pKa, etc..., where the activities cannot be understood in terms of elementary ligand-receptor interactions.

Observations with no Explanations¶
SAR observations may be uninterpretable. This may arise from (1) insufficient informational content of the data; (2) the simplicity of our models; (3) our ignorance of other factors; (4) an experimental error. Until additional data allow for an acceptable solution to emerge, the SAR remain unclear. Look at the data below and try to explain the SAR; imagine new molecules to be synthesized that might contribute to providing an answer. A possible rationale is proposed in the associated sub-page.

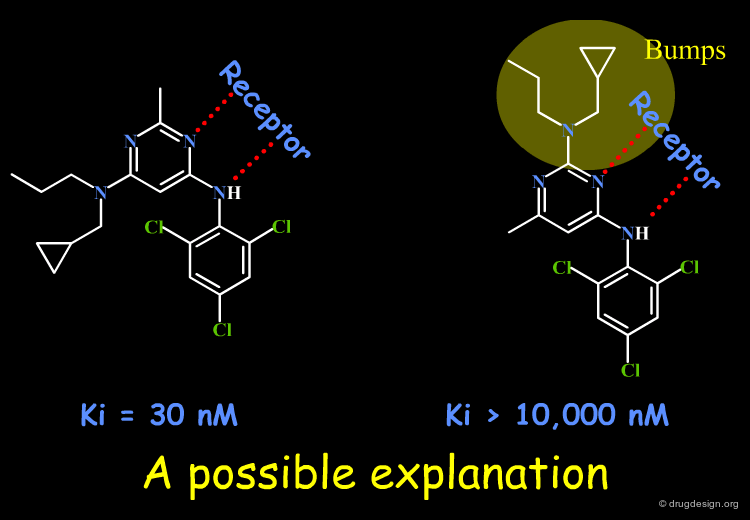
articles
Design and Synthesis of a Series of Non-Peptide High-Affinity Human Corticotropin-Releasing Factor1 Receptor Antagonists Chen Chen, Raymond Dagnino, Jr., Errol B. De Souza, Dimitri E. Grigoriadis, Charles Q. Huang, Kyung-Il Kim, Zhengyu Liu, Terry Moran, Thomas R. Webb, Jeffrey P. Whitten, Yun Feng Xie, and James R. McCarthy J. Med. Chem. 39 1996 10.1021/jm960149e
book
Wermuth CG Rational Approaches to Drug Design Prous Science 2001
Local Changes and Global Consequences¶
SAR analyses are based on the principle of independence: a local change is treated as not affecting the rest of the structure; however this is not always true. Sometimes a simple change in a molecule may completely change the property of the entire molecule (e.g. conformation) or its binding interaction (e.g. binding orientation). This is illustrated by a number of examples in the following pages.

Small Modification that Substantially Alters the Geometry¶
In the example shown below, a small structural modification, such as the replacement of the N-H by a N-CH3 group, completely alters the conformational preference of the original molecule. This effect should not be overlooked when interpreting the biological activities of the two analogs.

Small Modification that Interferes with Other Interactions¶
In the example represented here, the introduction of a methyl group leads to the complete loss of biological activity. The methyl group hampers the formation of an essential hydrogen bond interaction that existed upon binding of the original compound. The impact of the local change that was made can only be understood by looking at the molecule as a whole.


articles
218th ACS National Meeting: New Orleans, Louisiana August 22-26, 1999 Pechulis AD Albany Molecular Research, Inc. Technical Report 4(14) 2000
Modification that Alters the Binding Orientation¶
By analogy with ATP, Olomoucine (an ATP competitive inhibitor of the protein kinase CDK2) was expected to have the same binding mode as found for ATP. However, X-ray studies demonstrated an entirely different binding mode for it (analyzed in detail in the chapter structure-based drug design: analysis). Such a drastic effect should not be overlooked when interpreting the biological activities of adenine-based inhibitors (this is analyzed in detail in the chapter structure-based drug design: analysis).

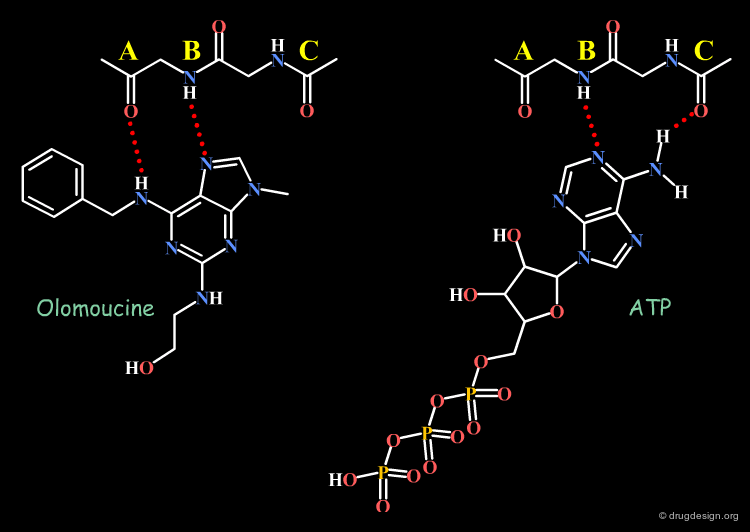
articles
Multiple modes of ligand recognition: crystal structures of cyclin-dependent protein kinase 2 in complex with ATP and two inhibitors, olomoucine and isopentenyladenine Schulze-Gahmen U, Brandsen J, Jones HD, Morgan DO, Meijer L, Vesely J, Kim SH Proteins 22 1995
Structural basis of inhibitor selectivity in MAP kinases Wang Z, Canagarajah BJ, Boehm JC, Kassisa S, Cobb MH, Young PR, Abdel-Meguid S, Adams JL, Goldsmith EJ Structure 6 1998
Pdb
Human Cyclin-Dependent Kinase 2, PDB entry: 1HCL
The Complex Structure Of The Map Kinase Erk2/Olomoucine, PDB entry: 4ERK
Change that Alters a Characteristic of the Substance¶
A small alteration of a structure may introduce substantial changes in specific physico-chemical properties, with repercussions in the biological properties, which can be even more pronounced in-vivo. For example, the replacement of a carbon atom by an oxygen, or the change in the number of carbon atoms in a molecule can affect many properties of the substance. Examples of changes in pKa and logP are presented in the following pages.
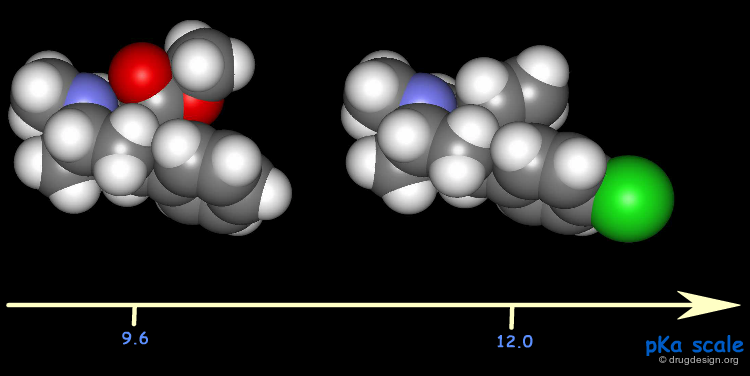
pKa¶
The difference in the pKa values of cocaine analogs was called upon to explain the differences in their binding potencies: a high potency is associated with a high pKa. Note the considerable changes that occurred in the pKa values.

articles
Conformational, Aqueous Solvation, and pKa Contributions to the Binding and Activity of Cocaine, WIN 35 065-2, and the WIN Vinyl Analog. Yang B, Wright J, Eldefrawi ME, Pou S and MacKerell AD HJ. Am. Chem. Soc 116 1994
Chemistry, Design, and Structure-Activity Relationship of Cocaine Antagonists Singh S Chem. Rev 100 2000
LogP¶
When changes are introduced in a structure, their impact in the global characteristics of the modified compounds must be considered. The example below illustrates a change in logP that is not negligible.

Non Additivity of Biological Effects¶
Knowing the biological effects of mono-structural changes in a series does not make it possible to predict the biological activity of an analog containing multiple changes simultaneously. SAR often rely on the principle of additivity of the biological properties: however this is not always true. In the following pages some examples of non-additivity of biological effects are presented.

Additivity in Steroids: Norethisterone¶
The norethisterone (NET) steroid was used as a starting point for SAR. The following single modifications were considered and led to compounds with increased potencies.

articles
Influence of the substitution of 11-methylene, Δ15(/sup}, and/or 18-methyl groups in norethisterone on receptor binding, transactivation assays and biological activities in animals Deckers GH, Schoonen WGEJ and Kloosterboer HJ J. of Steroid Biochem. and Mol. Biolog 74 2000
Additivity of Biological Effects¶
The introduction of two modifications simultaneously, leads to compounds with biological activities that are about the sum of those observed with single changes.
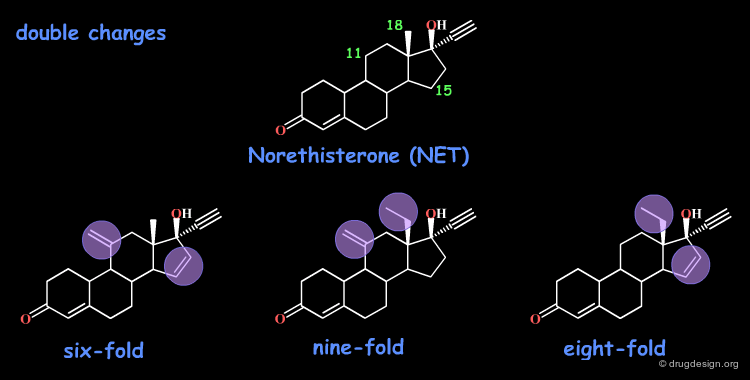
Non Additivity of Biological Effects¶
A compound twelve times more potent would be expected, when the three modifications are introduced simultaneously. In fact, the molecule is only nine times more potent than the initial reference steroid. In this example the biological effects are not simply additive.

Furanyl Example: Non-Additivity¶
In the example below the substitution of one furanyl group, improves the biological activities, but the bi-substituted analog is found to be less active.

articles
Design and synthesis of novel PPARalpha/gamma/delta triple activators using a known PPARalpha/gamma dual activator as structural template Mogensen JP, Jeppesen L, Bury PS, Pettersson I, Fleckner J, Nehlin J, Frederiksen KS, Albrektsen T, Din N, Mortensen SB, Svensson LA, Wassermann K, Wulff EM, Ynddal L abd Sauerberg P Bioorg. Med. Chem. Lett. 13 2003
Phenoxypyrimidines: Non Additivity¶
In the phenoxypyrimidine series, it has been observed that the mono-methyl substituted analogs at C-2 or at C-4 have biological activities close to that of the unsubstituted compound. On the contrary, the analog with the two methyl groups, is three times more potent.

articles
Phenoxypyrimidine inhibitors of p38alpha kinase: synthesis and statistical evaluation of the p38 inhibitory potencies of a series of 1-(piperidin-4-yl)-4-(4-fluorophenyl)-5-(2-phenoxypyrimidin-4-yl) imidazoles Boehm JC, Bower MJ, Gallagher TF, Kassis S, Johnson SR and Adams JL Bioorg. Med. Chem. Lett 11 2001
Being Trapped with Poor Biological Activities¶
The danger of being trapped in a series with poor biological activities is high when the receptor site is not known. When a lead is chosen, it is useful to make sure that the corresponding scaffold does not contain structural elements that are detrimental to the biological activities. It is worth spending some time addressing this issue before embarking on an SAR optimization program with a bad scaffold.

The Dioxobenzothiazole Scaffold¶
The dioxobenzothiazole scaffold has been the object of SAR studies, in a project aiming at the discovery of CDK4 kinase inhibitors. Starting with the unsubstituted derivative having an IC50 of 6 µM, a series of analogs were prepared, whose activities remained in the same micromolar range.

articles
5-Arylamino-2-methyl-4,7-dioxobenzothiazoles as inhibitors of cyclin-dependent kinase 4 and cytotoxic agents Ryu CK, Kang HY, Lee SK, Nam KA, Hong CY, Ko WG and Lee BH Bioorg. Med. Chem. Lett 10 2000
Origin of the Problem¶
Now that the interactions of inhibitors with the CDK4 kinase are well understood, it is possible to interpret the origin of the problem as being due to steric and polar repulsions between the nitrogen atom of the thiazole and the carbonyl of the hinge. Note that another scaffold containing an N-H group in this position could be ideal for improving the biological activities!
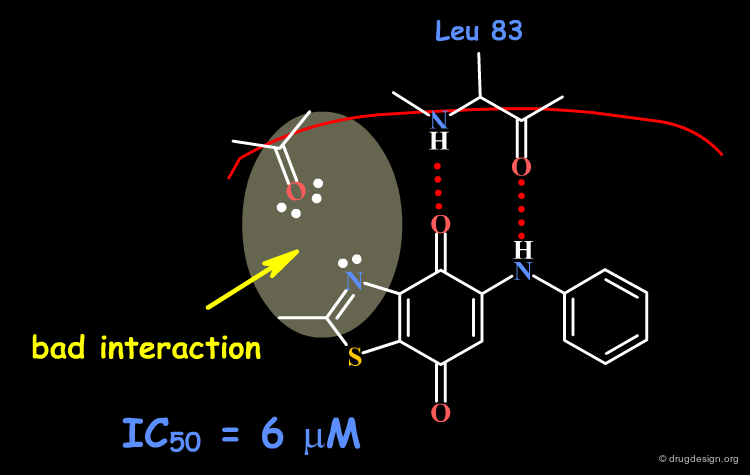
The Combinatorial Era¶
Our combinatorial chemistry-HTS era tends to generate what is sometimes called a "combinatorial chaos". Indeed computerized SAR programs are developed to handle this situation however the danger is to work with black-box-tools where the exact physico-chemical content diminishes. The real issue in SAR is to convert the data into useful knowledge that can be further exploited. Statistically-based programs cannot replace the creative and time consuming work needed to analyze SAR and extract this knowledge.

Example of Good Exploitation of SAR Complexity¶
The Banyu Story with the Urea Structure¶
Following a structure-based strategy, the Banyu group identified a hit (Cdk4 inhibitors project) having an urea moiety with an IC50 of 44 µM. The hit was simplified (ortho substituent removed) in order to derive further knowledge based on SAR analyses. In the following pages an outline of the important steps that enabled successful exploitation of SAR is presented.

articles
Structure-Based Generation of a New Class of Potent Cdk4 Inhibitors: New de Novo Design Strategy and Library Design Honma T, Hayashi K, Aoyama T, Hashimoto N, Machida T, Fukasawa K, Iwama T, Ikeura C, Ikuta M, Suzuki-Takahashi I, Iwasawa Y, Hayama T, Nishimura S and Morishima H J. Med. Chem 44 2001
A Novel Approach for the Development of Selective Cdk4 Inhibitors: Library Design Based on Locations of Cdk4 Specific Amino Acid Residues Honma T, Yoshizumi T, Hashimoto N, Hayashi K, Kawanishi N, Fukasawa K, Takaki T,Ikeura C, Ikuta M, Suzuki-Takahashi I, Hayama T, Nishimura S and Morishima H J. Med. Chem 44 2001
Importance of the Entire Urea Moiety¶
The complete loss of biological activity observed for the analog in which the anilino nitrogen atom is replaced by a carbon shows that the entire urea moiety is important for biological activities.

articles
Structure-Based Generation of a New Class of Potent Cdk4 Inhibitors: New de Novo Design Strategy and Library Design Honma T, Hayashi K, Aoyama T, Hashimoto N, Machida T, Fukasawa K, Iwama T, Ikeura C, Ikuta M, Suzuki-Takahashi I, Iwasawa Y, Hayama T, Nishimura S and Morishima H J. Med. Chem 44 2001
A Novel Approach for the Development of Selective Cdk4 Inhibitors: Library Design Based on Locations of Cdk4 Specific Amino Acid Residues Honma T, Yoshizumi T, Hashimoto N, Hayashi K, Kawanishi N, Fukasawa K, Takaki T,Ikeura C, Ikuta M, Suzuki-Takahashi I, Hayama T, Nishimura S and Morishima H J. Med. Chem 44 2001
Bioactive Conformation?¶
Based on modeling analyses the Banyu group suspected that the binding of the compounds involved the N-H and the carbonyl by forming two hydrogen bonds with the protein, in a bidentate manner. This hypothesis requires a "cis" conformation for the amide fragment concerned.

articles
Structure-Based Generation of a New Class of Potent Cdk4 Inhibitors: New de Novo Design Strategy and Library Design Honma T, Hayashi K, Aoyama T, Hashimoto N, Machida T, Fukasawa K, Iwama T, Ikeura C, Ikuta M, Suzuki-Takahashi I, Iwasawa Y, Hayama T, Nishimura S and Morishima H J. Med. Chem 44 2001
A Novel Approach for the Development of Selective Cdk4 Inhibitors: Library Design Based on Locations of Cdk4 Specific Amino Acid Residues Honma T, Yoshizumi T, Hashimoto N, Hayashi K, Kawanishi N, Fukasawa K, Takaki T,Ikeura C, Ikuta M, Suzuki-Takahashi I, Hayama T, Nishimura S and Morishima H J. Med. Chem 44 2001
Design of Compounds with a Cis Conformation¶
The aromatic phenyl was replaced by a pyridine, for all possible positions of the nitrogen atom (ortho, meta, para). The increased activity observed only for the 2-pyridyl isomer was interpreted as due to the increased population of the cis conformation (favored by an internal hydrogen bond).

articles
Structure-Based Generation of a New Class of Potent Cdk4 Inhibitors: New de Novo Design Strategy and Library Design Honma T, Hayashi K, Aoyama T, Hashimoto N, Machida T, Fukasawa K, Iwama T, Ikeura C, Ikuta M, Suzuki-Takahashi I, Iwasawa Y, Hayama T, Nishimura S and Morishima H J. Med. Chem 44 2001
A Novel Approach for the Development of Selective Cdk4 Inhibitors: Library Design Based on Locations of Cdk4 Specific Amino Acid Residues Honma T, Yoshizumi T, Hashimoto N, Hayashi K, Kawanishi N, Fukasawa K, Takaki T,Ikeura C, Ikuta M, Suzuki-Takahashi I, Hayama T, Nishimura S and Morishima H J. Med. Chem 44 2001
Good Exploitation of the SAR Analyses¶
The SAR analyses were further exploited and led to the discovery of very potent and selective compounds such as 15a. In summary, the major steps in this project were (1) discovery of a hit; (2) importance of the urea moiety; (3) modeling the probable bioactive conformation; (4) analogs favoring the cis conformation; (5) lead optimization.
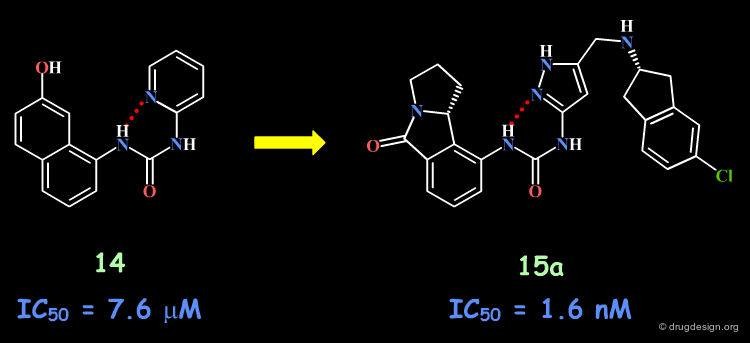
articles
Structure-Based Generation of a New Class of Potent Cdk4 Inhibitors: New de Novo Design Strategy and Library Design Honma T, Hayashi K, Aoyama T, Hashimoto N, Machida T, Fukasawa K, Iwama T, Ikeura C, Ikuta M, Suzuki-Takahashi I, Iwasawa Y, Hayama T, Nishimura S and Morishima H J. Med. Chem 44 2001
A Novel Approach for the Development of Selective Cdk4 Inhibitors: Library Design Based on Locations of Cdk4 Specific Amino Acid Residues Honma T, Yoshizumi T, Hashimoto N, Hayashi K, Kawanishi N, Fukasawa K, Takaki T,Ikeura C, Ikuta M, Suzuki-Takahashi I, Hayama T, Nishimura S and Morishima H J. Med. Chem 44 2001
Copyright © 2024 drugdesign.org