Case Studies of Docking in Drug Discovery¶
Info
Practical examples are discussed in some detail in this chapter to illustrate the application of docking in virtual screening, lead hopping, structure-activity relationships and the elucidation of modes of action.
Number of Pages: 52 (±1 hours read)
Last Modified: September 2008
Prerequisites: None
Case Study 1 : Pyrimidin-4-yl-ureas for Kinase Inhibition¶
Inhibitor Active on Several Protein Kinases¶
In an attempt to find a scaffold for protein kinase inhibition, a Novartis team developed a scaffold morphing strategy. They discovered a pyrimidin-4-yl urea structure that inhibited some protein kinases as indicated below.
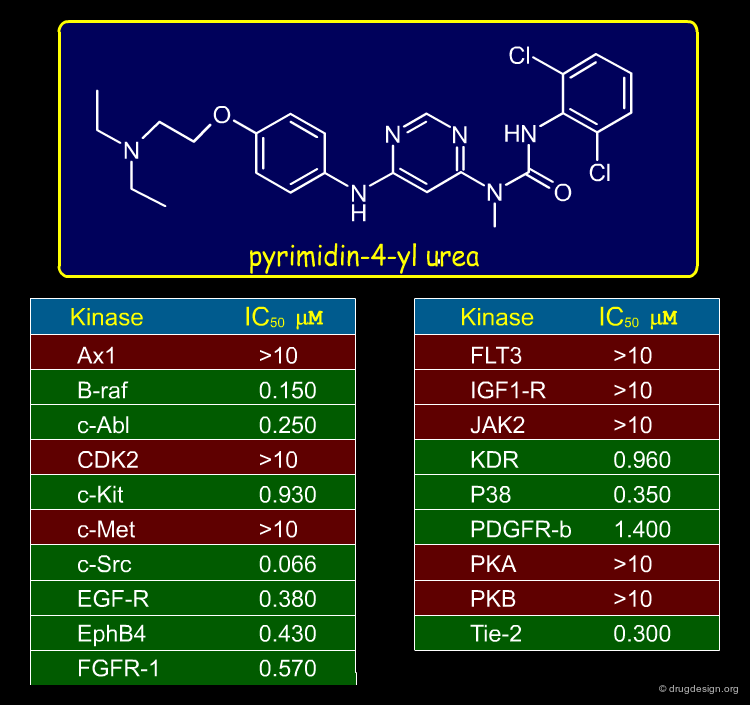
articles
Entry into a new class of protein kinase inhibitors by pseudo ring design P. Furet, G. Caravatti, V. Guagnano, M. Lang, T. Meyer and J. Schoepfer Bioorganic and Medicinal Chemistry Letters 18 2008
Structural Determinants for the Activity¶
Modeling studies were conducted to get a better grasp of the selectivity of the pyrimidin-4-yl urea. The molecule was docked in the ATP pocket of the c-Abl kinase, one of the protein kinases that was inhibited by that molecule. The 3D model revealed very tight interactions with the hydrophobic back pocket of the ATP binding site, in particular with the threonine gate keeper residue.

articles
Entry into a new class of protein kinase inhibitors by pseudo ring design P. Furet, G. Caravatti, V. Guagnano, M. Lang, T. Meyer and J. Schoepfer Bioorganic and Medicinal Chemistry Letters 18 2008
Correlation with the Volume of Gate Keeper Residue¶
The existence of tight binding of the pyrimidin-4-yl urea with the c-Abl threonine gatekeeper prompted the Novartis team to analyze the nature of the gatekeeper residues in the different kinases. They found a good correlation between the biological activities and the volume of the gate keeper. Protein kinases with a gatekeeper whose volume is larger than 125 Å3 cannot be active.

articles
Entry into a new class of protein kinase inhibitors by pseudo ring design P. Furet, G. Caravatti, V. Guagnano, M. Lang, T. Meyer and J. Schoepfer Bioorganic and Medicinal Chemistry Letters 18 2008
Outcome of this Study¶
This example illustrates how modeling analyses enabled the identification of a simple molecular determinant for interpreting the selectivity of a molecule. The selectivity of the pyrimidin-4-yl urea molecule was found to be directly correlated with the volume occupied by the gate keeper residue located in the back pocket of the ATP binding site.
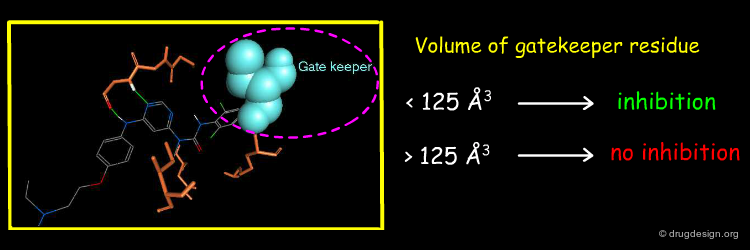
articles
Entry into a new class of protein kinase inhibitors by pseudo ring design P. Furet, G. Caravatti, V. Guagnano, M. Lang, T. Meyer and J. Schoepfer Bioorganic and Medicinal Chemistry Letters 18 2008
Case Study 2 : Inhibition of CHK1¶
The CHK1 Kinase¶
Chk1 is a kinase which contributes to the "G2 to M" transition in the cell-cycle. If DNA is damaged, an active Chk1 allows healthy cells to arrest in G2 and repair their DNA. Many mainstream anti-cancer agents act by damaging the DNA of cancer cells, leading to cell-death by apoptosis after mitosis. Therefore, the goal is to inhibit Chk1 with drugs in these cells to prevent them from repairing their DNA. The X-ray structure of the kinase domain has been solved, which provided impetus for a structure-based approach to the discovery of Chk1 inhibitors. The binding modes of several inhibitors discovered by virtual screening and analyzed by X-ray crystallography and modelling analyses are presented in the following pages.

Author
Nicolas Foloppe Vernalis Ltd. Cambridge, United Kingdom
The Indazole Series¶
To acquire experience with the system, Nicolas Foloppe and a Vernalis team studied the binding modes of publicly disclosed indazole series of Chk1 inhibitors.
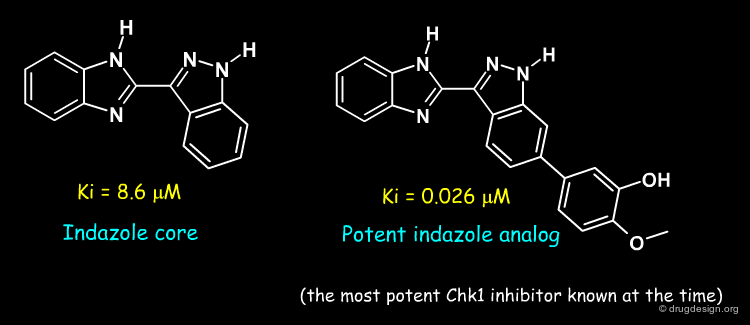
Author
Nicolas Foloppe Vernalis Ltd. Cambridge, United Kingdom
articles
Targeting Serine/Threonine Protein Kinase B/Akt and Cell-cycle Checkpoint Kinases for Treating Cancer Qun Li and Gui-Dong Zhu Curr. Topics Med. Chem 2 (9) 2002
Binding Mode of the Indazole Core¶
The series' indazole core was docked in the site, forming the conventional hydrogen-bonds with the conserved hinge. This provided a firm basis to model other compounds in the series and learn from their SAR, as shown in the next pages.

Author
Nicolas Foloppe Vernalis Ltd. Cambridge, United Kingdom
articles
Identification of a buried pocket for potent and selective inhibition of Chk1: Prediction and verification Nicolas Foloppe, Lisa M. Fisher, Geraint Francis, Rob Howes, Peter Kierstan and Andrew Potter Biorg. Med. Chem. 14 2006
Binding Modes of the Potent Indazole Analog¶
Modelling of the most potent compound in the indazole series, with a methoxy-phenol substituent, proved very informative. It showed that the methoxy-phenol fit sterically very well in a buried pocket which is otherwise filled with 3 water molecules. Burial of the methoxy-phenol, its excellent complementarity to the protein, and displacement of the waters explained the associated gain in potency. Therefore, this analysis identified this buried pocket as an important target to enhance potency against Chk1. The modelled binding mode was subsequently confirmed by crystallography by the Vernalis team (see rotatable view).

Author
Nicolas Foloppe Vernalis Ltd. Cambridge, United Kingdom
articles
Identification of a buried pocket for potent and selective inhibition of Chk1: Prediction and verification Nicolas Foloppe, Lisa M. Fisher, Geraint Francis, Rob Howes, Peter Kierstan and Andrew Potter Biorg. Med. Chem. 14 2006
Pocket may Help for Selectivity¶
Selectivity is an important issue when targeting a kinase for medicinal chemistry. A structural overlay of several different kinases around the buried pocket and the methoxy-phenol showed that the side-chains around this pocket varied across kinases. Modelling and experiments confirmed that this pocket is important for selectivity as well as potency.

Author
Nicolas Foloppe Vernalis Ltd. Cambridge, United Kingdom
articles
Identification of a buried pocket for potent and selective inhibition of Chk1: Prediction and verification Nicolas Foloppe, Lisa M. Fisher, Geraint Francis, Rob Howes, Peter Kierstan and Andrew Potter Biorg. Med. Chem. 14 2006
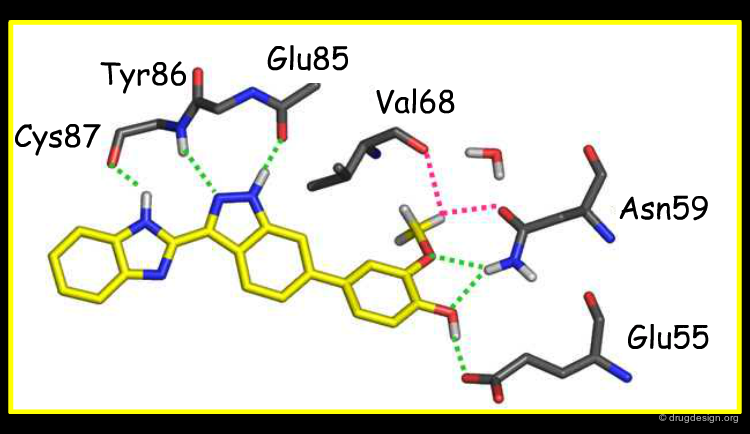
Overlay with Other Chk1 Inhibitors¶
The view below shows an overlay of some known Chk1 inhibitors. The indazole series (red) fills the buried pocket in Chk1, at the bottom of which is Asn59. The other series do not take advantage of this pocket. Therefore, there are opportunities to design novel and more specific Chk1 inhibitors.
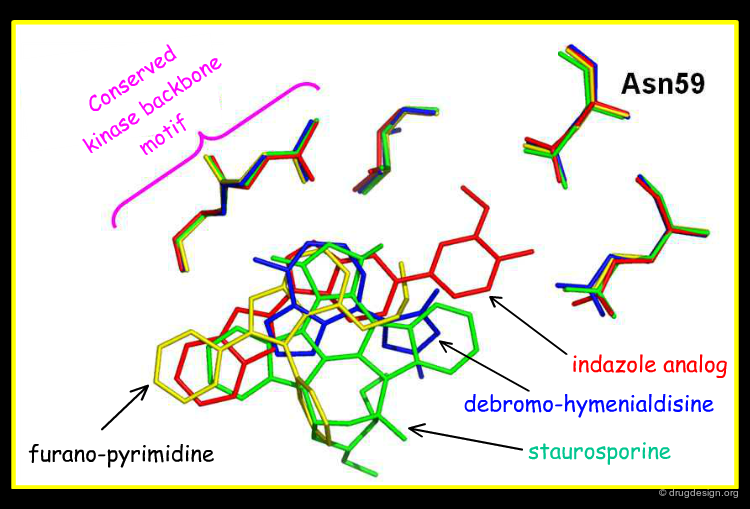
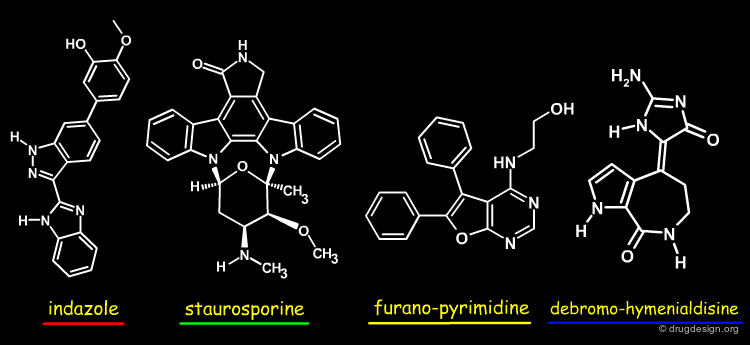
Author
Nicolas Foloppe Vernalis Ltd. Cambridge, United Kingdom
articles
Identification of a buried pocket for potent and selective inhibition of Chk1: Prediction and verification Nicolas Foloppe, Lisa M. Fisher, Geraint Francis, Rob Howes, Peter Kierstan and Andrew Potter Biorg. Med. Chem. 14 2006
Structure-Based Screening of Chk1 Inhibitors¶
The modelling analyses on indazole analogs helped better identify the requirements for binding to the Chk1 kinase. The Vernalis team then searched for new inhibitors. A virtual screening methodology was deployed and started with a large electronic catalog of commercially available compounds. Compounds with undesirable properties (extremes of polarity, reactive functionalities) were filtered before docking. This left about 0.7 million compounds which were docked in high-throughput. Computational and manual filtering of the docking pauses led to assay a relatively small number of compounds. These finally yielded nine new diverse scaffolds of Chk1 inhibitors.

Author
Nicolas Foloppe Vernalis Ltd. Cambridge, United Kingdom
articles
Identification of chemically diverse Chk1 inhibitors by receptor-based virtual screening Nicolas Foloppe, Lisa M. Fisher, Rob Howes, Andrew Potter, Alan G. S. Robertson and Allan E. Surgenor Biorg. Med. Chem. 14 2006
Hits Identified by Virtual Screening¶
The chemical structures of the nine new scaffolds of Chk1 inhibitors discovered by docking in the ATP-binding site of Chk1 are shown below. A key point is that these scaffolds are diverse, arguably reflecting the strength of receptor-based docking as compared to a more similarity orientated ligand-based approach. All the inhibitors are ATP-competitive Chk1 inhibitors, which inhibit Chk1 at low micromolar concentrations of the compound.

Author
Nicolas Foloppe Vernalis Ltd. Cambridge, United Kingdom
articles
Identification of chemically diverse Chk1 inhibitors by receptor-based virtual screening Nicolas Foloppe, Lisa M. Fisher, Rob Howes, Andrew Potter, Alan G. S. Robertson and Allan E. Surgenor Biorg. Med. Chem. 14 2006
X-Ray Structures of Four Virtual Screening Hits¶
X-ray structures of four of the newly discovered Chk1 inhibitors bound to this target were obtained. Each compound hydrogen-bonds the conserved kinase hinge, as expected. These X-ray binding modes confirmed some, but not all, of the docking modes obtained during the high-throughput virtual screening (not an unusual situation).



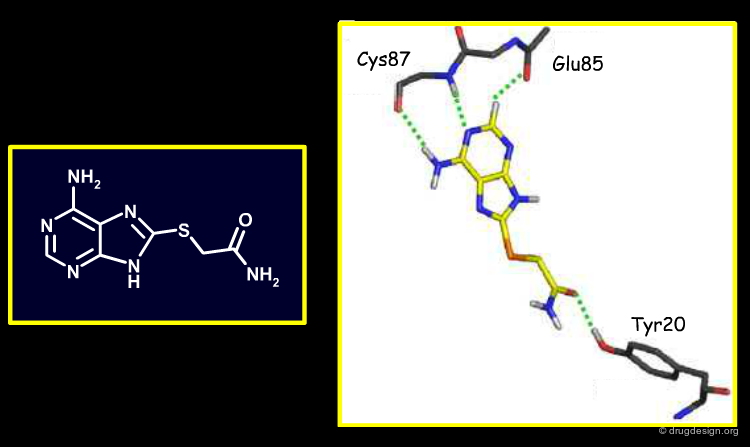
Author
Nicolas Foloppe Vernalis Ltd. Cambridge, United Kingdom
articles
Identification of chemically diverse Chk1 inhibitors by receptor-based virtual screening Nicolas Foloppe, Lisa M. Fisher, Rob Howes, Andrew Potter, Alan G. S. Robertson and Allan E. Surgenor Biorg. Med. Chem. 14 2006
Binding Modes Predicted for Other Five Hits¶
Since X-ray crystallography failed for the five remaining scaffolds, their precise binding modes (BM) were predicted by rigorous docking studies. These models emphasize the formation of hydrogen-bonds with the kinase hinge; however, due to the symmetry of this hydrogen-bonding motif, some of these docking modes could be flipped by 180 degrees relative to the hinge. These are only working hypotheses that are very helpful when devising an initial medicinal-chemistry strategy; they can then be adjusted in the light of new data.

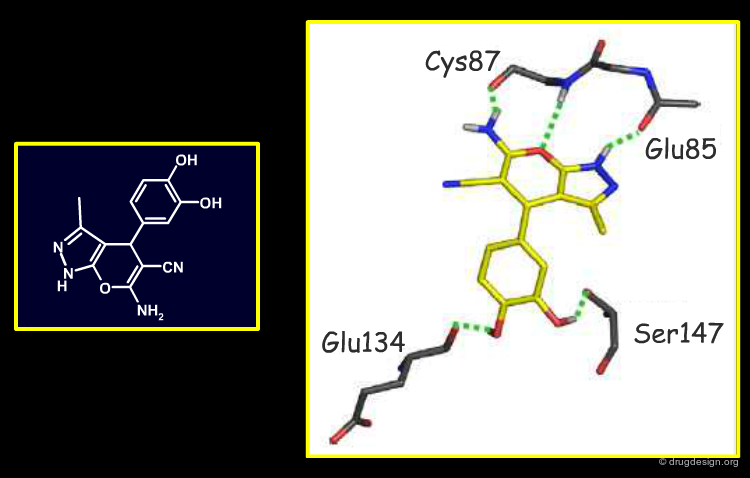


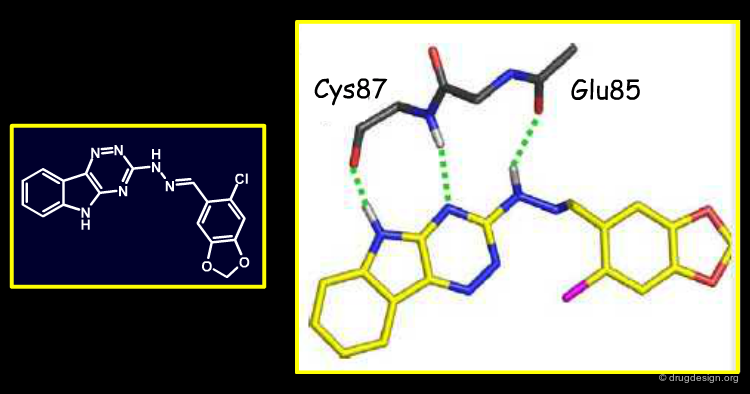
Author
Nicolas Foloppe Vernalis Ltd. Cambridge, United Kingdom
articles
Identification of chemically diverse Chk1 inhibitors by receptor-based virtual screening Nicolas Foloppe, Lisa M. Fisher, Rob Howes, Andrew Potter, Alan G. S. Robertson and Allan E. Surgenor Biorg. Med. Chem. 14 2006
Outcome of this Study¶
The structural models generated in this study are helpful in providing concrete starting points to inform further inhibitor design. Since a limited number of alternative sensible docking modes could be found for each of the scaffolds, this suggests that structure-based ligand design is a realistic prospect for several of these ligands.

Author
Nicolas Foloppe Vernalis Ltd. Cambridge, United Kingdom
articles
Identification of a buried pocket for potent and selective inhibition of Chk1: Prediction and verification Nicolas Foloppe, Lisa M. Fisher, Geraint Francis, Rob Howes, Peter Kierstan and Andrew Potter Biorg. Med. Chem. 14 2006
Identification of chemically diverse Chk1 inhibitors by receptor-based virtual screening Nicolas Foloppe, Lisa M. Fisher, Rob Howes, Andrew Potter, Alan G. S. Robertson and Allan E. Surgenor Biorg. Med. Chem. 14 2006
Case Study 3 : Thrombin Inhibitors¶
Two Methods of Virtual Screening¶
Two approaches are currently used in virtual screening (VS). The first one, called structure-based virtual screening, exploits knowledge of the 3D structure of the target protein, and the second one, called ligand-based scoring, evaluates whether molecules mimic the 3D and physicochemical properties of a known active reference compound. A hybrid approach combining the two methods and applied on thrombin inhibitors is presented in the following pages.

Author
Leslie A. Kuhn and Maria I. Zavodszky Department of Biochemistry and Molecular Biology, Michigan State University, USA
Combining Structure-Based and Ligand-Based VS¶
Leslie Kuhn and her group at the Michigan State University explored how to take advantage of the benefits of both structure-based docking (evaluating how well a compound complements the structure and chemistry of the active site) and ligand-based scoring (evaluating whether a compound shares features with known inhibitors) by combining them. This was tested in the context of a search for thrombin inhibitors, and the results are presented in the following pages.

Author
Leslie A. Kuhn and Maria I. Zavodszky Department of Biochemistry and Molecular Biology, Michigan State University, USA
articles
** ** Zavodszky, M.I., Rohatgi, A., Van Voorst, J., Yan, H., Kuhn, L.A Journal of Molecular Recognition
2008
Screening Protocol¶
The screening protocol considered for this study is outlined in the figure below. In the first phase, the structure-based screening method is applied, based on the SLIDE method introduced by the Kuhn group (2002). In the second phase, the docked compounds are ranked by comparing each candidate molecule to that of the potent inhibitor PPACK, using a ligand-based 3D electrostatic similarity scoring method (EON).
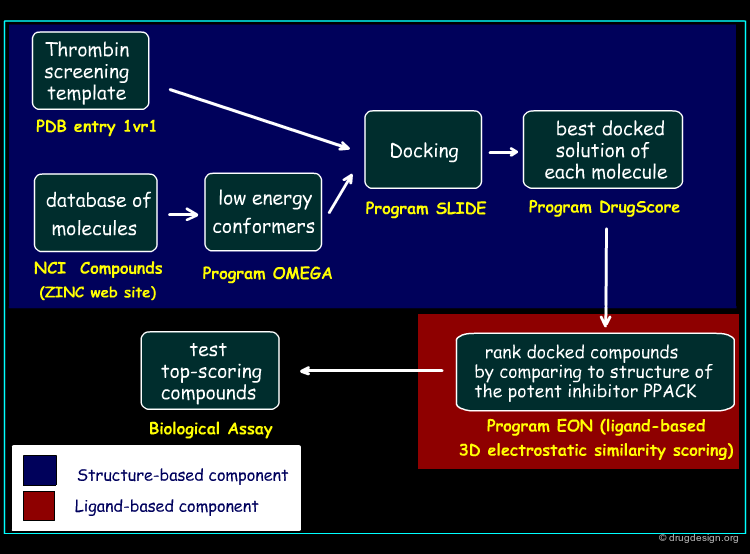
Author
Leslie A. Kuhn and Maria I. Zavodszky Department of Biochemistry and Molecular Biology, Michigan State University, USA
articles
** ** Zavodszky, M.I., Rohatgi, A., Van Voorst, J., Yan, H., Kuhn, L.A Journal of Molecular Recognition
2008
Distilling the essential features of a protein surface for improving protein-ligand docking, scoring, and virtual screening Zavodszky, M.I., Sanschagrin, P.C., Korde, R.S., Kuhn, L.A. J. Comput. Aided Mol. Des 16 2002
Knowledge-based scoring function to predict protein-ligand interactions Gohlke, H., Hendlich, M., Klebe, G. J. Mol. Biol 295 2000
Steps of the Docking Treatment¶
The steps involved in the docking (SLIDE program) are summarized in the diagram below. This is a flexible docking method where both the protein and ligand side-chains are treated as flexible entities; they mutually adapt to resolve van der Waals overlaps until good fit is achieved.

Author
Leslie A. Kuhn and Maria I. Zavodszky Department of Biochemistry and Molecular Biology, Michigan State University, USA
articles
Screening a peptidyl database for potential ligands to proteins with side-chain flexibility Schnecke, V., Swanson, C.A., Getzoff, E.D., Tainer, J.A., Kuhn, L.A. Proteins 33 1998
Atlas of Side-Chain and Main-Chain Hydrogen Bonding McDonald and Thornton
Distilling the essential features of a protein surface for improving protein-ligand docking, scoring, and virtual screening Zavodszky, M.I., Sanschagrin, P.C., Korde, R.S., Kuhn, L.A. J. Comput. Aided Mol. Des 16 2002
Specificity Pockets in Thrombin¶
The active site of thrombin is visualized in the view below. The small spheres (template points) represent positions of favored interactions for inhibitor candidates to make, based on the protein structure alone; they were generated by the SLIDE method (Zavodszky et al., 2002). The template points are colored according to their type: green for hydrophobic, red for acceptor, blue for donor, and purple for donor and/or acceptor points. Specificity pockets are labeled; the S2 pocket is behind the tryptophan side chain at top.
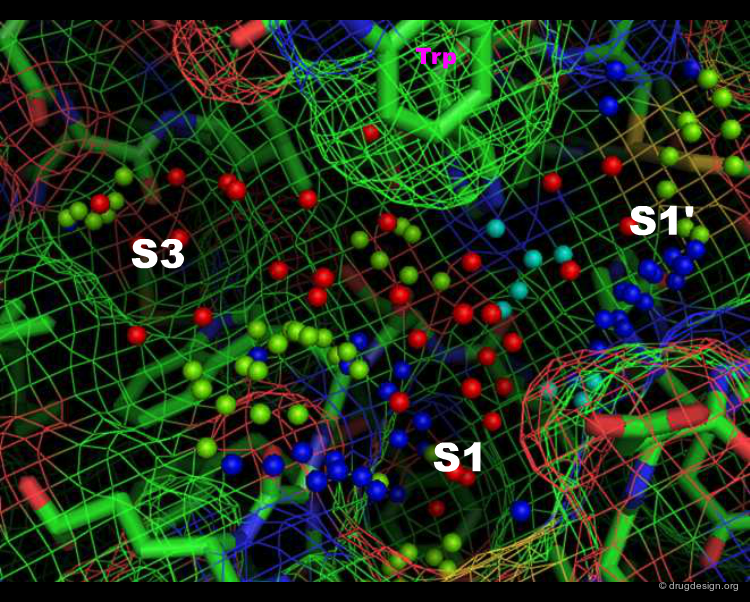
Author
Leslie A. Kuhn and Maria I. Zavodszky Department of Biochemistry and Molecular Biology, Michigan State University, USA
articles
Distilling the essential features of a protein surface for improving protein-ligand docking, scoring, and virtual screening Zavodszky, M.I., Sanschagrin, P.C., Korde, R.S., Kuhn, L.A. J. Comput. Aided Mol. Des 16 2002
Development of the Hybrid Approach¶
The thrombin target (PDB 1vr1) and 66777 unique compounds from the NCI database were screened. Based on good fit in the thrombin active site and matching of at least three of the thrombin template points, 7618 inhibitor candidates were identified for further evaluation. The best docked orientation for each compound was selected based on the structure-based scoring method, DrugScore. The 3D electrostatic similarity of each of the docked inhibitor candidates was then compared with the crystallographic orientation of the potent thrombin inhibitor, PPACK, using EON software. The EON scores were used to rank the compounds for experimental testing.
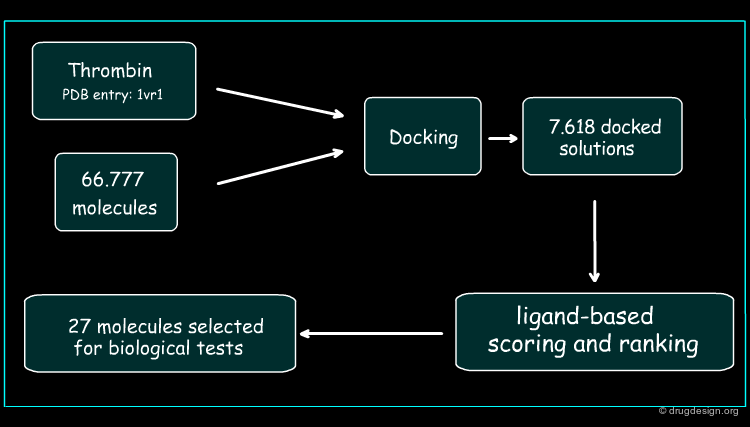
Author
Leslie A. Kuhn and Maria I. Zavodszky Department of Biochemistry and Molecular Biology, Michigan State University, USA
articles
** ** Zavodszky, M.I., Rohatgi, A., Van Voorst, J., Yan, H., Kuhn, L.A Journal of Molecular Recognition
2008
Knowledge-based scoring function to predict protein-ligand interactions Gohlke, H., Hendlich, M., Klebe, G. J. Mol. Biol 295 2000
Inhibition Assays of Top-Scoring Compounds¶
27 of the NCI Plated Compounds were obtained for biological testing, and the corresponding results are shown below. The assays were repeated in the presence of detergent, showing no evidence of inhibition by aggregation (promiscuous inhibition) for any of these compounds. Molecule 1 was found to have a sub-micromolar IC50.
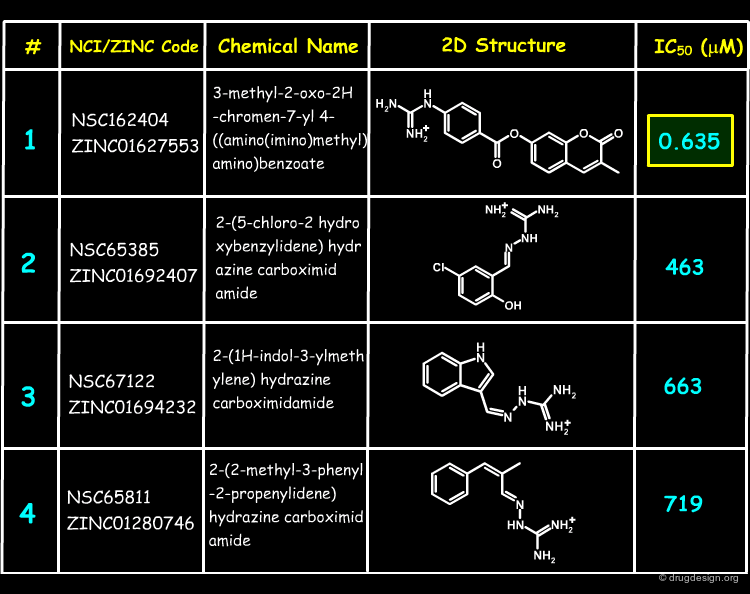
Author
Leslie A. Kuhn and Maria I. Zavodszky Department of Biochemistry and Molecular Biology, Michigan State University, USA
articles
** ** Zavodszky, M.I., Rohatgi, A., Van Voorst, J., Yan, H., Kuhn, L.A Journal of Molecular Recognition
2008
A Common Mechanism Underlying Promiscuous Inhibitors from Virtual and High-Throughput Screening Susan L. McGovern, Emilia Caselli, Nikolaus Grigorieff and Brian K. Shoichet J. Med. Chem 45 2002
Analysis of the Binding Mode of Compound 1¶
Below is shown the predicted binding mode of compound 1 (in white tubes) as compared to the binding mode of the PPACK inhibitor (green tubes). Note that although PPACK was the reference inhibitor used to rank NCI compounds for ligand-based similarities, compound 1 incorporates new thrombin interactions. Compound 1 and PPACK have strong volumetric and electrostatic overlap in the S1 pocket, resulting in the high EON score between the two.

Author
Leslie A. Kuhn and Maria I. Zavodszky Department of Biochemistry and Molecular Biology, Michigan State University, USA
articles
** ** Zavodszky, M.I., Rohatgi, A., Van Voorst, J., Yan, H., Kuhn, L.A Journal of Molecular Recognition
2008
Binding Mode Compared with Known Inhibitors¶
This view represents the binding mode of compound 1 compared to 42 known thrombin inhibitors from crystal structures in the PDB; it shows that this compound makes more extensive contacts in the S1' pocket than other inhibitors.

Author
Leslie A. Kuhn and Maria I. Zavodszky Department of Biochemistry and Molecular Biology, Michigan State University, USA
articles
** ** Zavodszky, M.I., Rohatgi, A., Van Voorst, J., Yan, H., Kuhn, L.A Journal of Molecular Recognition
2008
What was Learned in this Test Study ?¶
This test study led to the discovery of a sub-micromolar inhibitor forming unique interactions with thrombin that can be used as a starting point for further optimization. The results show that ligand-based scoring can be a successful alternative to scoring protein-ligand interactions when prioritizing compounds for experimental testing.

Author
Leslie A. Kuhn and Maria I. Zavodszky Department of Biochemistry and Molecular Biology, Michigan State University, USA
articles
** ** Zavodszky, M.I., Rohatgi, A., Van Voorst, J., Yan, H., Kuhn, L.A Journal of Molecular Recognition
2008
Analyzing Top Ranked Compounds¶
The Kuhn group compared the top 100 compounds ranked by the ligand similarity scoring method EON to the top 100 compounds ranked by DrugScore, which measures protein-ligand interactions, and found there were no compounds in common. Considering the top 100 compounds from the protein-ligand interaction scoring function built into SLIDE, AffiScore/N, only 3 compounds were in common with the EON top-100 list. Furthermore, the AffiScore/N and DrugScore top 100 lists had no compounds in common.
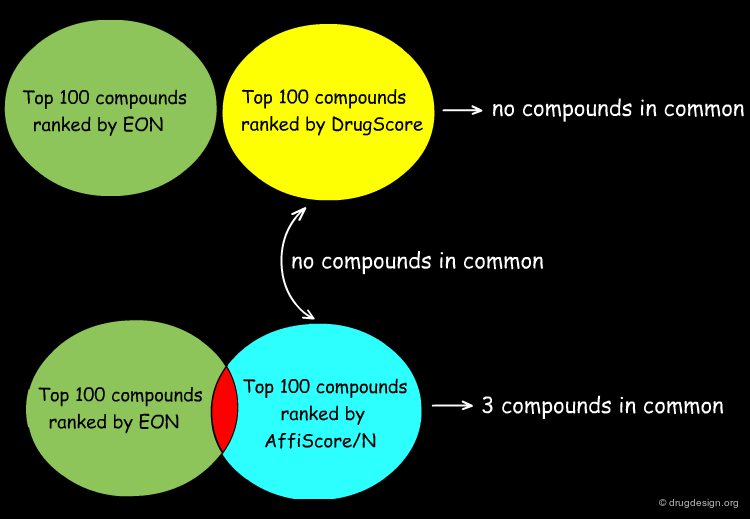
Author
Leslie A. Kuhn and Maria I. Zavodszky Department of Biochemistry and Molecular Biology, Michigan State University, USA
articles
** ** Zavodszky, M.I., Rohatgi, A., Van Voorst, J., Yan, H., Kuhn, L.A Journal of Molecular Recognition
2008
Limitations of Scoring Functions¶
Thus, even though all three scoring functions have proven effective in selecting good dockings across diverse protein complexes, there are still considerable differences between them when it comes to prioritization of compounds. A practical approach for dealing with this challenge is to test several different scoring functions for their ability to correctly rank the known inhibitors for the target considered.

Author
Leslie A. Kuhn and Maria I. Zavodszky Department of Biochemistry and Molecular Biology, Michigan State University, USA
articles
** ** Zavodszky, M.I., Rohatgi, A., Van Voorst, J., Yan, H., Kuhn, L.A Journal of Molecular Recognition
2008
Case Study 4 : Salicylamide Renin Inhibitor¶
Search for New Scaffold in Renin Inhibition¶
CGP-54061A is a potent non-peptide renin inhibitor that was discovered using a structure-based approach (see the aliskiren story). This antihypertensive agent was designed to mimic a reference peptide-like inhibitor, as indicated in the figure below. Here we present the achievements of Cohen and Maibaum at Novartis in their effort to discover a new series with a scaffold that differed from that of CGP54061A.

articles
Design and Synthesis of Novel, Fully Non-Peptide Transition State Mimic Renin Inhibitors bearing an O-alkyl Substitued Salicylamide (P3-sp-P3)-Moiety with High Oral in vivo Potency J. Maibaum, N.C. Cohen, J. Rahuel, Ch. Schnell, H-P. Baum, P. Rigollier, W. Schilling, J. Wood XVth Meeting of the European Federation of Medicinal Chemistry
Edinburg, Sept. 6-10, 1998
3D Analyses¶
The analyses started with the X-ray data of the reference peptide (in purple in the view) and the non-peptidic inhibitor. The isopropyl group of CGP-54061A mimics the cyclohexyl of the peptide and the methoxybenzene mimics the Phe residue of the peptide. Initially, the benzene of CGP-54061A was intended to mimic the Phe residue of the peptide but the 3D mimicry was not optimal, and this is why a methoxy group was added in para, in order to better overlap with the Phe residue of the peptide.

Preferred Location of Phenyl Ring in Pocket P3¶
Looking at the 3D mimicry of the peptide and CGP-54061A and based on the fact that both molecules have an IC50 of 1 nM, the authors attempted to determine the preferred location for a phenyl ring in the P3 pocket.

Docking Experiment¶
To find the answer, the phenyl-dialkoxy of the CGP-54061A inhibitor was disconnected from the rest of the molecule, and the remaining two disconnected parts were then docked simultaneously - each one being free to bind optimally to the enzyme. Note that the para methoxy group was removed from the docking simulation (originally added to better overlap with the Phe residue).

Results of the Docking¶
The docking simulation revealed that the aromatic ring of the phenyl-dialkoxy moved approximately 2Å away from the other moiety, and reached the location occupied by the phenyl ring of the Phe residue of the reference peptide molecule (grey). In the absence of connectivity constraints, the preferred location of the phenyl ring of CGP-54061A in P3 corresponds to that occupied by the peptide.

Search for an Optimal Spacer¶
Knowing the docked preferred position of the two fragments in the active site of the enzyme, the next issue was to assemble the two pieces to form one unique molecule.
![]()
The Salicylamide Lead¶
On the screen, Cohen and Maibaum searched for spacers that would assemble the two disconnected parts in 3D. The carboxamidomethylene (CO-NH-CH2) spacer was selected as the best solution; the first molecule that was prepared proved to be active at the nanomolar level, and led to a new salicylamide family of renin inhibitors.

articles
Design and Synthesis of Novel, Fully Non-Peptide Transition State Mimic Renin Inhibitors bearing an O-alkyl Substitued Salicylamide (P3-sp-P3)-Moiety with High Oral in vivo Potency J. Maibaum, N.C. Cohen, J. Rahuel, Ch. Schnell, H-P. Baum, P. Rigollier, W. Schilling, J. Wood XVth Meeting of the European Federation of Medicinal Chemistry
Edinburg, Sept. 6-10, 1998
Predictions Confirmed by X-Ray Study¶
The predicted bound conformation of the salicylamide lead was confirmed by subsequent X-ray determination of its complex with renin. In the view below, two amino-acids of the catalytic site were removed to enable the entire inhibitor to be seen.
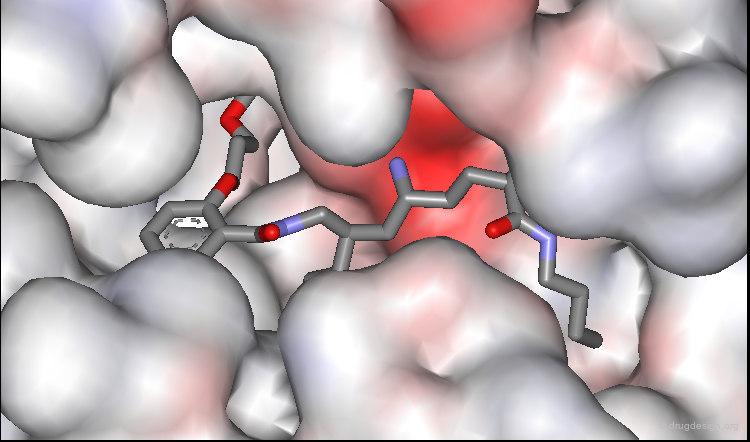
Browser of Salicylamide Inhibitor¶

Optimization of the Salicylamide Series¶
The initial lead compound 1 was then optimized by addressing both the in-vitro binding (with the purified enzyme and in the presence of plasma) and the in-vivo binding (marmoset plasma renin). Compound 4 proved to be highly potent in-vitro and in-vivo, water soluble and orally active.

articles
Design and Synthesis of Novel, Fully Non-Peptide Transition State Mimic Renin Inhibitors bearing an O-alkyl Substitued Salicylamide (P3-sp-P3)-Moiety with High Oral in vivo Potency J. Maibaum, N.C. Cohen, J. Rahuel, Ch. Schnell, H-P. Baum, P. Rigollier, W. Schilling, J. Wood XVth Meeting of the European Federation of Medicinal Chemistry
Edinburg, Sept. 6-10, 1998
Summary¶
Starting with an initial lead compound, molecular modeling analyses and docking treatments could be used to optimally position the phamacophoric fragments of the lead compound. The assembly of these fragments in their new position led to the design and discovery of the salicylamide series, an entirely new family of renin inhibitors.

Lead Hopping¶
This example illustrates how docking treatments can be efficiently exploited to create new scaffolds by lead hopping.

Case Study 5 : Inhibition of Human Neutrophil Elastase¶
Inhibition of Human Neutrophil Elastase¶
Human neutrophil elastase (HNE), a serine protease found in the azurophilic granules of neutrophils, is involved in many inflammatory diseases such as rheumatoid arthritis, pulmonary emphysema, delayed wound healing and psoriasis. Hence, inhibition of human neutrophil elastase is an interesting therapeutic approach as regards these inflammatory diseases. Up to now there are no elastase inhibitor medications on the market for therapy. Looking for new human neutrophil elastase inhibitors, natural compounds can be a valuable source and sesquiterpene lactones were found by the Merfort group to have such inhibitory activities. Docking analyses were used to elucidate their mode of action. This is presented in the following pages.

Author
Irmgard Merfort and Bettina Siedle Institute of Pharmaceutical Sciences, Department of Pharmaceutical Biology and Biotechnology, University of Freiburg, Germany
articles
Natural compounds as inhibitors of human neutrophil elastase Siedle B., Hrenn A., Merfort I Planta Medica 73 2007
Sesquiterpene lactones as inhibitors of human neutrophil elastase Siedle, B., Cisielski, S., Murillo, R., Loeser, B., Castro, V., Klaas, C.A., Hucke, O., Labahn, A., Melzig, M.F., Merfort, I Bioorganic and Medical Chemistry 10 2002
Sesquiterpene Lactones¶
Sesquiterpene Lactones (SLs) are terpenoids that are the active constituents of a variety of medicinal plants used in traditional medicine for the treatment of inflammatory diseases. More than 4000 different structures are known which can be divided into four major groups according to their carbocyclic skeleton. Each of these groups can be divided into subgroups.


Author
Irmgard Merfort and Bettina Siedle Institute of Pharmaceutical Sciences, Department of Pharmaceutical Biology and Biotechnology, University of Freiburg, Germany
articles
Sesquiterpene lactones as inhibitors of human neutrophil elastase Siedle, B., Cisielski, S., Murillo, R., Loeser, B., Castro, V., Klaas, C.A., Hucke, O., Labahn, A., Melzig, M.F., Merfort, I Bioorganic and Medical Chemistry 10 2002
Sesquiterpene lactones as taxonomic characters in the Asteraceae Seaman F.C The Botanical Review 48 1982
Quantitative structure - activity relationship of sesquiterpene lactones as inhibitors of the transcription factor NF-kappaB Siedle, B., Garcia-Pineres, A.J., Murillo, R., Schulte-Moenting, J., Castro, V., Ruengeler, P., Klaas, C.A., Da Costa, F., Kisiel, W., Merfort, I J. Med. Chem 47 2004
Studies on 17 Sesquiterpene Lactones¶
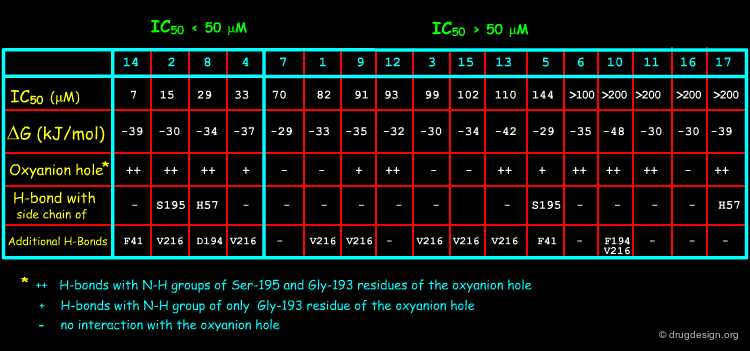
For their investigations the Merfort group selected seventeen sesquiterpene lactones representing all major skeletal classes of SLs. They differ from each other both in their molecular geometry and in the number of α,β-unsaturated carbonyl structures. Only four of them (14, 2, 8 and 4) had an IC50 < 50 µM in the elastase assay, the others required a high concentration for the 50% inhibition.

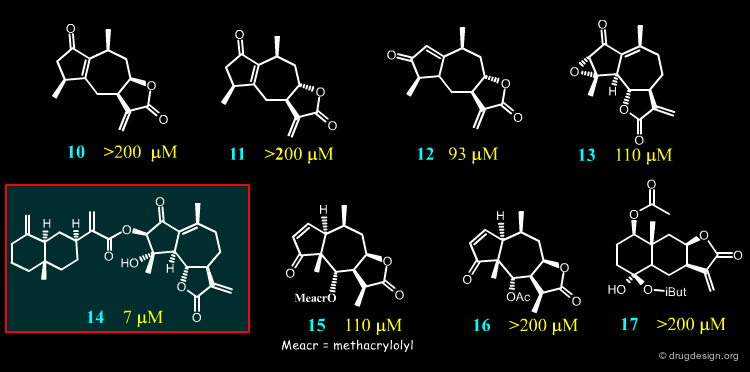

Author
Irmgard Merfort and Bettina Siedle Institute of Pharmaceutical Sciences, Department of Pharmaceutical Biology and Biotechnology, University of Freiburg, Germany
articles
Sesquiterpene lactones as inhibitors of human neutrophil elastase Siedle, B., Cisielski, S., Murillo, R., Loeser, B., Castro, V., Klaas, C.A., Hucke, O., Labahn, A., Melzig, M.F., Merfort, I Bioorganic and Medical Chemistry 10 2002
Docking Studies¶
In an attempt to probe the activities and the mode of action of sesquiterepene lactones such as 14, 2, 8 and 4, docking studies were conducted on Human Neutrophil Elastase.

Author
Irmgard Merfort and Bettina Siedle Institute of Pharmaceutical Sciences, Department of Pharmaceutical Biology and Biotechnology, University of Freiburg, Germany
Docking Protocol¶
The X-ray structure of HNE was obtained from the PDB. Its binding site was determined with the molecular modeling package WhatIf. Low-energy conformations of the SLs were generated using the molecular modeling package Hyperchem. Docking calculations were carried out with the FlexX program. The predicted protein-ligand complexes were optimized and ranked according to the empirical scoring function ScreenScore, which estimates the binding free energy of the ligand receptor complex. The best placement refers to the ligand receptor complex with the lowest binding free energy.

Author
Irmgard Merfort and Bettina Siedle Institute of Pharmaceutical Sciences, Department of Pharmaceutical Biology and Biotechnology, University of Freiburg, Germany
articles
Structure of Human Neutrophil elastase in complex with a peptide chloromethyl ketone inhibitor at 1.84-A resolution (1HNE) Navia, M. A., McKeever, B. M., Springer, J. P., Lin, T., Williams, H. R., Fluder, E. M., Dorn, C. P., Hoogsteen, K. Proceedings of the National Academy of Science USA 86 1989
WHAT IF: A molecular modeling and drug design program Vriend, G. Journal of Molecular Graphics 8 1990
A Fast Flexible Docking Method using an Incremental Construction Algorithm (FlexX) Rarey, M., Kramer, B., Lengauer, T., Klebe, G. Journal of Molecular Biology 261 1996
Detailed analysis of scoring functions for virtual screening (ScreenScore) Stahl, M., Rarey, M. Journal of Medicinal Chemistry 44 2001
Results of the Docking Studies¶
The best active compounds (2, 4, 8 and 14) were always found the ones that all anchored to the oxyanion hole (Ser-195 and Gly-193), or at least partly, by hydrogen bonds. This anchoring may prevent the natural substrate to bind to the catalytic triad. The docking studies revealed no obvious correlation with the free energies estimated by ScreenScore, but can explain the inhibitory activity within structurally related SLs, as illustrated for the melampolides 2, 3 and 4 on the following pages.
Author
Irmgard Merfort and Bettina Siedle Institute of Pharmaceutical Sciences, Department of Pharmaceutical Biology and Biotechnology, University of Freiburg, Germany
articles
Sesquiterpene lactones as inhibitors of human neutrophil elastase Siedle, B., Cisielski, S., Murillo, R., Loeser, B., Castro, V., Klaas, C.A., Hucke, O., Labahn, A., Melzig, M.F., Merfort, I Bioorganic and Medical Chemistry 10 2002
Elucidation of the Mode of Action¶
Altogether, docking studies help understand the activities and explain the mechanism of HNE inhibition of SLs: occupation of the oxyanion hole by H-bond anchorage and additional hydrogen bonds with other residues. The docking studies confirm the experimental results by showing that the inhibition is reversible. SLs do not covalently bind to the catalytic triad, thus differing from other elastase inhibitors with a lactone moiety.

articles
Quantitative structure - activity relationship of sesquiterpene lactones as inhibitors of the transcription factor NF-kappaB Siedle, B., Garcia-Pineres, A.J., Murillo, R., Schulte-Moenting, J., Castro, V., Ruengeler, P., Klaas, C.A., Da Costa, F., Kisiel, W., Merfort, I J. Med. Chem 47 2004
Kinetics and mechanism of human leukocyte elastase inactivation by ynenol lactones Copp, L.J. Kranz, A., Spencer, R.W Biochemistry 26 1987
Docking Results of Melampolides 2 and 4¶
The different activities of melampolides 2, 3 and 4 (2~4>3) can be explained by docking studies. Besides the binding to Gly-193 in the oxyanion hole, the two hydroxyl groups of SLs 2 and 4 enable a double anchoring of the molecule to the active site. These interactions may prevent the natural substrate from binding to the catalytic triad.



Author
Irmgard Merfort and Bettina Siedle Institute of Pharmaceutical Sciences, Department of Pharmaceutical Biology and Biotechnology, University of Freiburg, Germany
Docking Results of Podachaenin 14¶
The best inhibitor (14) is an ester having a hydrophobic bicyclic moiety that fills a major part of the furrow in which the physiological peptidic substrate is bound. This may explain its strong inhibitory activity.


Author
Irmgard Merfort and Bettina Siedle Institute of Pharmaceutical Sciences, Department of Pharmaceutical Biology and Biotechnology, University of Freiburg, Germany
Docking Results of Germacranolide 8¶
Docking studies with 8 confirm the importance of a hydroxy function near the lactone carbonyl for a good interaction within the active site.


Author
Irmgard Merfort and Bettina Siedle Institute of Pharmaceutical Sciences, Department of Pharmaceutical Biology and Biotechnology, University of Freiburg, Germany
Structural Determinants for Binding to HNE¶
Ligand binding docking calculations reveal that the occurrence of a carbonyl function together with a hydroxy group or two hydroxy groups at a certain distance from one another seem to be a prerequisite for the inhibitory activity of sesquiterpene lactones to human neutrophil elastase. This 3D pharmacophore may be used as a query in 3D database searching for novel inhibitors that are chemically unrelated to the structures of the sesquiterpene lactones.

Author
Irmgard Merfort and Bettina Siedle Institute of Pharmaceutical Sciences, Department of Pharmaceutical Biology and Biotechnology, University of Freiburg, Germany
articles
Sesquiterpene lactones as inhibitors of human neutrophil elastase Siedle, B., Cisielski, S., Murillo, R., Loeser, B., Castro, V., Klaas, C.A., Hucke, O., Labahn, A., Melzig, M.F., Merfort, I Bioorganic and Medical Chemistry 10 2002
Summary¶
Docking studies helped to explain the possible mode of inhibitory action of human neutrophil elastase inhibitors on sesquiterpene lactones and demonstrate their high potential in the search for lead structures in the development of human neutrophil elastase inhibitors starting from small natural molecules.

Author
Irmgard Merfort and Bettina Siedle Institute of Pharmaceutical Sciences, Department of Pharmaceutical Biology and Biotechnology, University of Freiburg, Germany
articles
Sesquiterpene lactones as inhibitors of human neutrophil elastase Siedle, B., Cisielski, S., Murillo, R., Loeser, B., Castro, V., Klaas, C.A., Hucke, O., Labahn, A., Melzig, M.F., Merfort, I Bioorganic and Medical Chemistry 10 2002
ADDITIONAL CASE STUDIES¶
Additional Case Studies¶

Copyright © 2024 drugdesign.org