Library Design¶
Info
The present chapter reviews the principles used in library design that can be applied for a variety of purposes ranging from the purchase of external libraries to the selection of molecules from a virtual or an existing pool of compounds, for testing them biologically. The subjects presented in this chapter include: methods for designing scaffolds, diversity and focused methodologies, how to measure molecular diversity, how to reduce a huge set of molecules, how to optimize the number of reagents and how integrate ADMET considerations in the design of a library.
Number of Pages: 135 (±3 hours read)
Last Modified: November 2008
Prerequisites: None
Introduction¶
Libraries of Molecules Prepared One by One¶
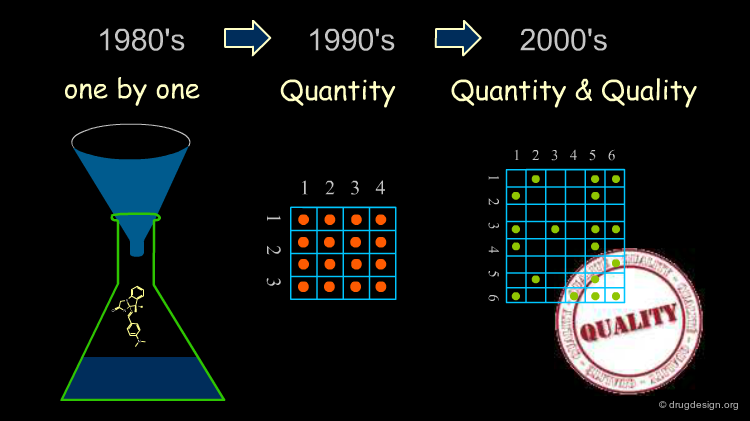
In the past a "library" consisted of a collection of molecules prepared one by one. The library contained molecules more for purposes of archiving, subsequent patent protection and multiple-project screening than for developing a comprehensive strategy designed to accelerate the discovery process for a project.
The Combinatorial Chemistry Boom¶
The combinatorial chemistry boom that emerged in the nineties enabled tens of thousands of compounds to be made in a single cycle, whereas only 50-70 compounds could be synthesized each year by a medicinal chemist using traditional methods. This breakthrough fueled new expectations based on the feeling that the increase in the number of molecules synthesized would lead to more chances to find a hit in the biological tests.

Initial Disapointments¶
Surprisingly, the increase in molecules generated by high throughput technologies did not substantially change the number of hits obtained over the last ten years, although the number of compounds synthesized and screened during this period increased by several orders of magnitude.

articles
Chemical space navigation in lead discovery Oprea TI Curr Opin Chem Biol 6 2002
The Quest of Quality¶
Early in the 2000's it became apparent that CombiChem and rapid high throughput synthesis capabilities were not just a game of numbers, but rather a matter of thorough design with an intelligent choice of compounds to be synthesized.
Chemical Diversity Space¶
It has been estimated that the total number of small organic molecules is about 10200, and this defines the entire space of all possible candidate molecules. With CombiChem technologies the medicinal chemist can design and synthesize many of them; however he must address important critical choices which are restricted by a number of factors including the scaffold limitations, reagents availability, synthetic feasibility, time frame, cost ceilings, laboratory capabilities etc...

Library Representation¶
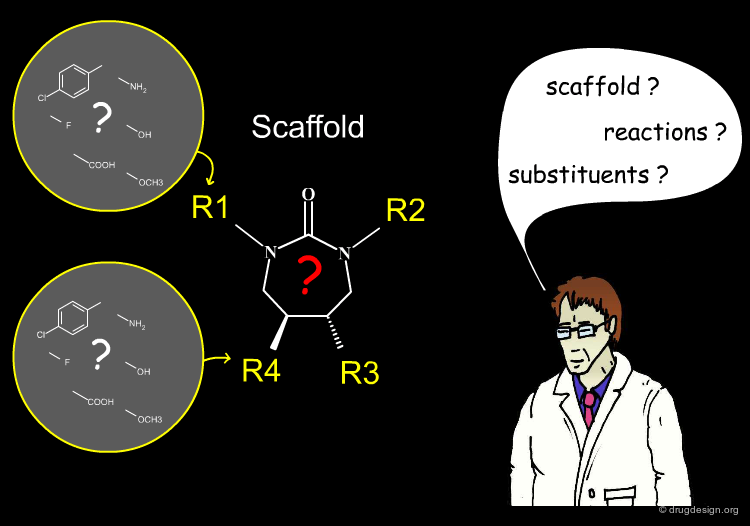
Most combinatorial chemical libraries can be represented as a fixed scaffold and a set of variable R-groups (commonly between 1 and 4). Each of these variable positions is filled by a set of fragments known as a substituent. In the table you can click either the headers (Ri) or the substituents.
Definition of a Virtual Library¶
The expression "virtual library" refers to the assembly of all the molecules that could potentially be made with a given scaffold using all possible reactants. For example, the following scaffold can generate one million products, the number of reagents available for R1, R2 and R3 being 200, 50 and 100, respectively. The number of products that can be generated far exceeds practical limits to be of use.

Scaffolds-Substituents-Reactions¶
An efficient library design program resides in the right combination of the following three features: scaffolds, substituents and reactions. In the following sections we discuss each of these issues in some detail.
The Basis of a Good Scaffold¶
Scaffold: the First Piece of a Complex Jigsaw Puzzle¶
The choice of the scaffold is the first major decision the medicinal chemist needs to make. It is the first element of a complex jigsaw puzzle that will lead the combinatorial chemistry effort towards success or to a dead end. The choice not only limits considerably the molecular space explored, but also predetermines many facets of the future lead.

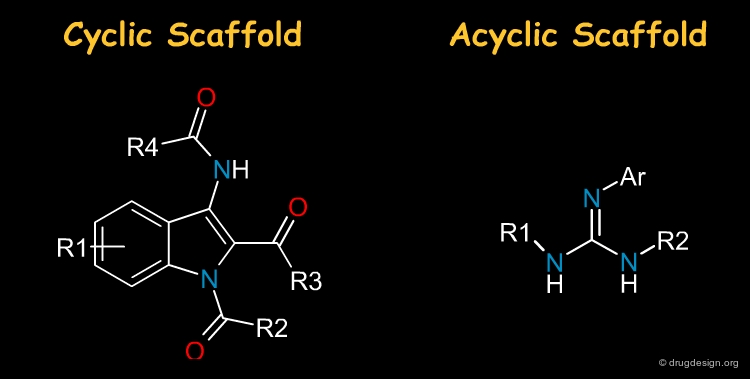
Cyclic and Acyclic Scaffold¶
Most drugs are made of heterocycles and it is not surprising that they are very often the first choice in library design. Nevertheless acyclic scaffolds are also important in drug discovery: the term "scaffold" should therefore be taken in a broad sense, i.e. including acyclic moeties.
articles
Recent developments in combinatorial organic synthesis A. Ganesan DDT 7 2002
The Scaffold, a Fuzzy Concept¶
A scaffold generally results from a reasoning comprising some fuzziness. Suppose for example that we want to develop a combinatorial chemistry program based on the structure of the hit shown below. What scaffold should be extracted? a simple imidazole moiety? a benzimidazole heterocycle? or should we include one or two of the substituents that were present in the initial hit?

The Scaffold Names¶
There are many designations for a scaffold. The most commonly used names are scaffold, template, structural fragment, core, building block, nucleus, moiety, skeleton etc...

Scaffold Requirements¶
The choice of a scaffold is based on thorough analyses assessing its capabilities with respect to multiple requirements. The ideal scaffold is expected to carry all the properties described below. In the following pages we discuss each of the different items appearing in this list.

Scaffold and ADME Properties¶
In the next sections we will show that the ADME-Tox properties of a lead are determined to a great extent by the properties of the scaffold. Once the scaffold is fixed, the modulation of these properties becomes restricted. It may be a challenge to substantially change the permeability or the solubility, for example. It is important for the physicochemical properties of the scaffold to carry favorable structural features so that compounds contained within the library constitute good starting points for further optimization.
articles
Drug-like properties and the causes of poor solubility and poor permeability. Lipinski CA. Journal of Pharmacological and Toxicological Methods. 44 2000
Geometrical Requirements¶
The geometrical, and sometimes also the conformational features of the scaffold, are of paramount importance. The scaffold must (1) not be exposed to risks of clashes with the binding sites; (2) present robust binding interactions and finally (3) it must present good directions for the substituents.

Avoid Clashes with the Receptor¶
The scaffold must not be exposed to the risk of clashes with the receptor, and this is the first requirement. Bumps with the receptor theoretically appear only with the substituents, not with the scaffold. The HIV-1 protease and Renin are both aspartic proteases with a similar catalytic site; in Renin however, the space available is much smaller. The cyclic urea is suited for HIV-1, whereas for Renin, only an acyclic scaffold can be envisaged.

Good Binding Interactions With the Scaffold¶
The second requirement for a scaffold is to contribute to binding. For example many scaffolds of kinase inhibitors present good anchorage capabilities with the formation of two hydrogen bonds, in a bidentate manner as illustrated below. These interactions are crucial for biological activities.

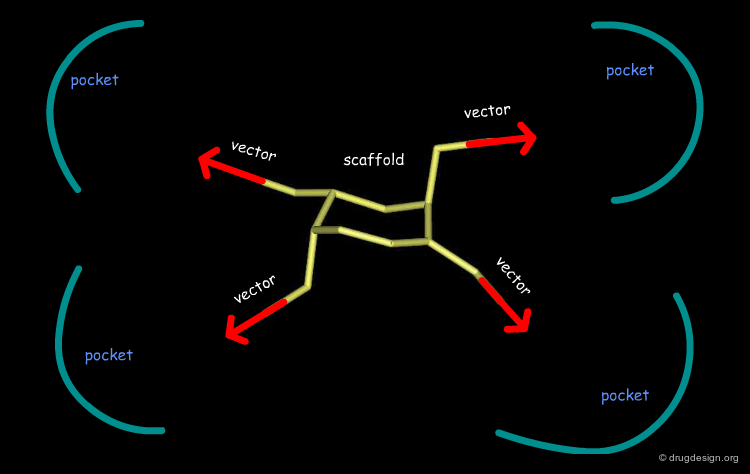
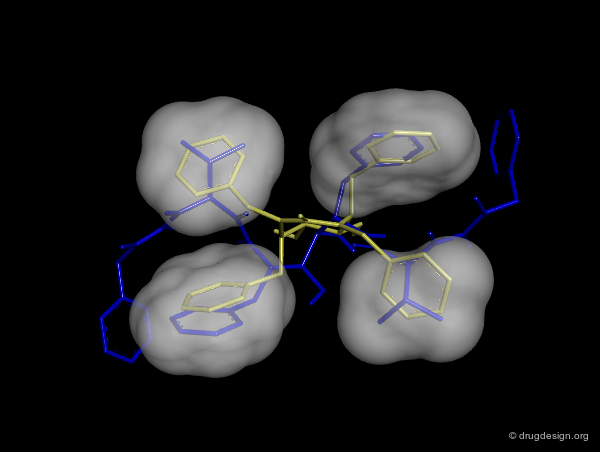
Good Vector Orientation¶
The third role of a scaffold is to hold the essential elements of the pharmacophore in the proper 3D geometrical orientation. In the following example the seven-membered ring scaffold carries the substituents in a good orientation that allows for favorable interactions with the receptor.
Patent Position and Novelty¶
R&D costs for a pharmaceutical company can reach $800 million to get a new chemical entity to the market, and effective protection of the molecules is therefore of the outmost importance. The scaffold is at the center of the intellectual property business and the chemical structures in the claim are examined for their novelty and patentability.

Problems of Scaffolds Patentability¶
The patentability of a scaffold is a difficult problem to solve. Very often the medicinal chemist is more or less submitted to the attraction of the "me-too-ism" or of the existence of a "hole" in a patent. Therefore some types of scaffolds repeatedly appear in a great number of patents.

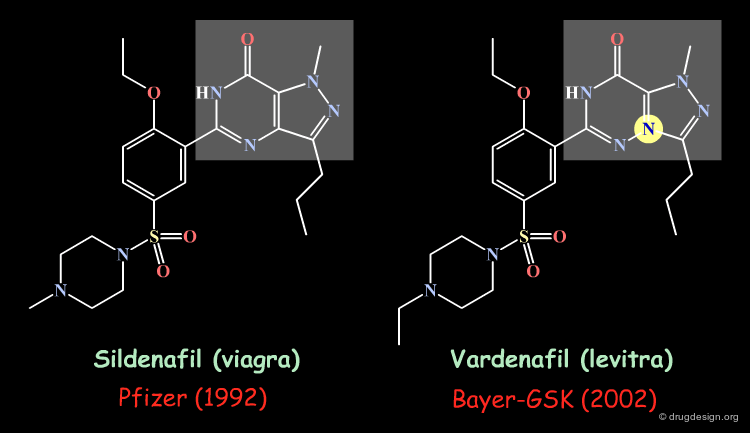
Patentable Scaffold¶
Bioisosteric transformation has been widely used to circumvent a particular patentability problem. This is illustrated below in the case of the sildenafil drug (viagra).
Validation of Novelty¶
Before embarking upon a costly development operation, it is important to make sure that the compounds can be effectively patented. The following illustrates some steps that can be taken to verify the patentability of a given scaffold. If the scaffold is not described in the literature (perhaps due to synthetic difficulties), this is a good opportunity for the company to reexamine its feasibility in the light of recent in-house technological advances.

Syntheses Amenable to Combinatorial Chemistry¶
The combinatorial synthesis approach of libraries of organic compounds for high throughput screening requires the availability of scaffolds amenable to combinatorial chemistry reactions, that also can be automated. This requires medicinal chemistry expertise and computational tools.

Bond-Formation for Array Synthesis Methods¶
In combinatorial chemistry the major challenge has been making carbon-carbon and carbon-heteroatom bond-forming reactions amenable to the methods of array synthesis. For example, ring closure metathesis, olefination and arylation of aromatic rings constitute some reaction classes suitable for combinatorial synthesis. Both solid phase and solution phase synthetic methods can be envisaged.

Ideal Scaffold: a Multi-Project Template¶
It is essential for a pharmaceutical company to reduce its R&D cost. One solution is to own one or two scaffolds that have the potential to interact with a large number of biological targets. These "master-scaffolds" allow the company to consider its R&D in a multi-project perspective. In a given sub-project, appropriate substituents can be introduced in the reference master-scaffold to generate a drug candidate that will achieve potency and selectivity for the disease under consideration.
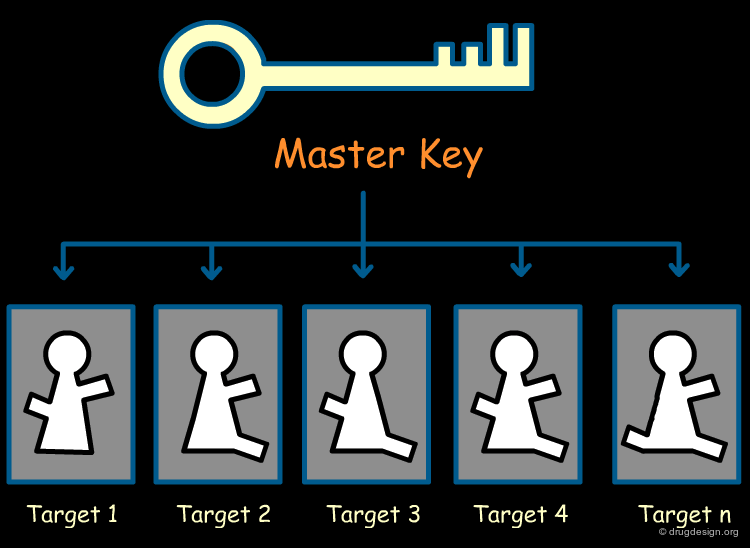

A Universal Spacer as a Master Key¶
A universal spacer is a core scaffold having structural characteristics that are compatible with the structures of a great number of biological targets. Starting with the initial proprietary master-scaffold, refined analogs are designed that exploit the particular 3D arrangement of the active site to achieve good binding interactions.
![]()
Diversity of the Space Covered by the Substituents¶
An important requirement for a scaffold to become a "master key" is to carry a good geometrical diversity in the virtual space of its substituents. This provides versatility and flexibility to the medicinal chemist, who decides where to introduce substituents that will adapt to the 3D requirements of many biological targets. The benzodiazepine structure, a sugar moiety or an indole scaffold (see below) are examples of templates with such diversity.

articles
Design and Quantitative Structure-Activity Relationship of 3-Amidinobenzyl-1H-indole-2-carboxamides as Potent, Nonchiral, and Selective Inhibitors of Blood Coagulation Factor Xa Hans Matter et al. J. Med. Chem 45 2002
Benzodiazepine Scaffold as a Spacer¶
The benzodiazepinedione scaffold is an example of a useful and versatile template that has been used as a core in a variety of therapeutic indications including anxiolytics, antiarrhythmics, vasopressin antagonists, HIV reverse transcriptase inhibitors, cholecystokinin antagonists etc.. This is an ideal moiety for high throughput synthetic strategies. Below is an illustration of how the indan-based structure of the SB209670 endothelin antagonist has been "translated" into the structure of a benzodiazepine mimic.

A Master Key Adapted for a Family¶
Another possibility of exploiting a scaffold in a multi-project approach consists of addressing a particular family of target proteins, where small modifications of the structure of a ligand can change the action of the molecule under consideration. A design technology can be produced that exploits the similarities and small differences of the 3D architectures of the proteins concerned, and enables the design of lead candidates for each of them, where potency and specificity are achieved in a rational way (possibly leading to drug candidates for different indication areas).

Sequence Similarity: Kinase Example¶
Proteins of the same family have similar sequences leading to active sites that are conserved. The following table illustrates an example of similarities and differences found in the signal transduction area (protein kinases). In the next pages we will illustrate how a reference scaffold can be modulated to specifically address different members of the family.
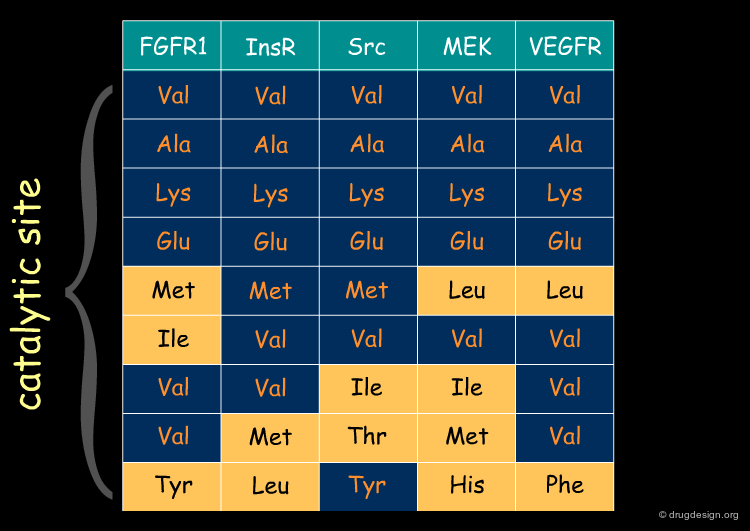

articles
Crystal structure of an angiogenesis inhibitor bound to the FGF receptor tyrosine kinase domain M. Mohammadi et al. EMBO J. 17 1998
Potent and Selective Inhibitors of the ABL-Kinase:Phenykamino-Pyrimidine (PAP) Derivatives Zimmermann J. Bioorganic and Medicinal Chemistry Letters 7 1997
Sequence Similarity in CDKi Sub-Class¶
The same type of pattern is even more pronounced within a given sub-class of proteins (in the example below: the cyclin dependent kinases CDKi).
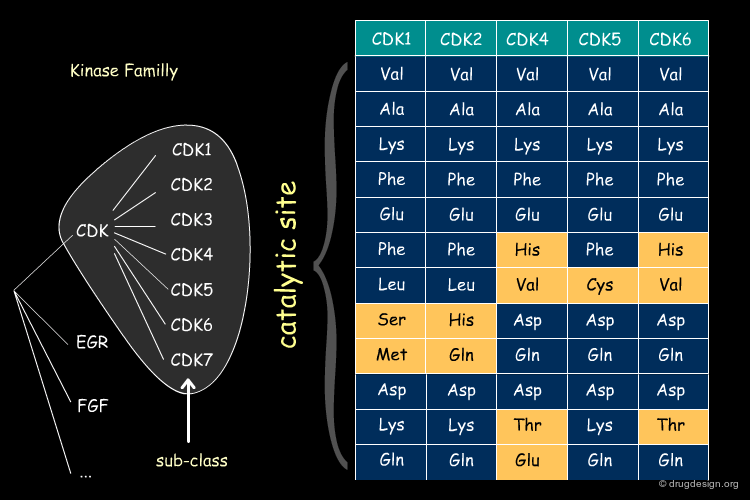
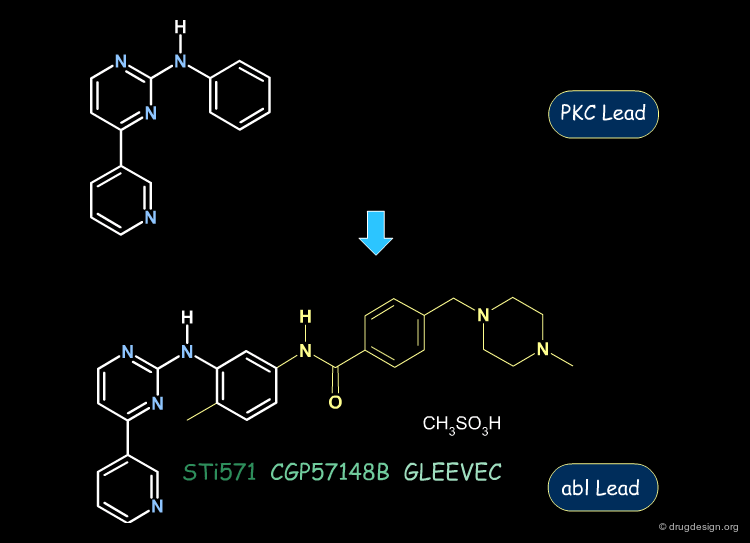
Gleevec: from PKC to Abl Inhibition¶
The following aminopyrimidine structure was initially explored for the inhibition of the PKC kinase. Then, further analogs and SAR analyses served to exploit this scaffold for the inhibition of the abl kinase. This enabled the discovery of Gleevec, the first protein kinase inhibitor ever introduced on the market.
Modulation with Simplified Staurosporine Scaffolds¶
Based on the structure of Staurosporine it has been possible to derive simplified Staurosporine analogs that were refined to generate lead compounds targeting the inhibition of various protein kinases such as PKC, EGF-R or PDGF inhibition.

Quinazoline Scaffold¶
Amino quinazolines represent an important class of scaffolds that has been exploited against different kinase targets. The example below illustrates how it is possible to shift from one target (EGF-R) to the other (CSF) with a simple structural modification (substitution of the amino nitrogen atom).

Scaffold Selection and Design¶
Methods for Designing a new Scaffold¶
Several methods are available to the medicinal chemist for designing new scaffolds. For simplicity of presentation we describe some of them: they appear in a sequential manner, but they are not independent: the design is generally a combination of several methods.

Small Modification of Known Scaffold¶
The most commonly used method for designing a new scaffold consists of modifying the structure of a known active compound. Many such examples are described in the chapter "Structure-Activity Relationships". In the illustration below, the structure of the imidazole derivative SB 203580 (p38 kinase inhibitor) was exploited to define another series with a pyrrole scaffold, such as L-167307.

articles
Pyrroles And Other Heterocycles as Inhibitors of P38 Kinase Stephen E. de Laszlo et al. Bioorganic and Medicinal Chemistry Letters 8 1998
2D Similarity Searching¶
Similarity searching finds molecules that contain the same general features as described in the query. The similarity is assessed, based on the comparison of 2D structural keys.

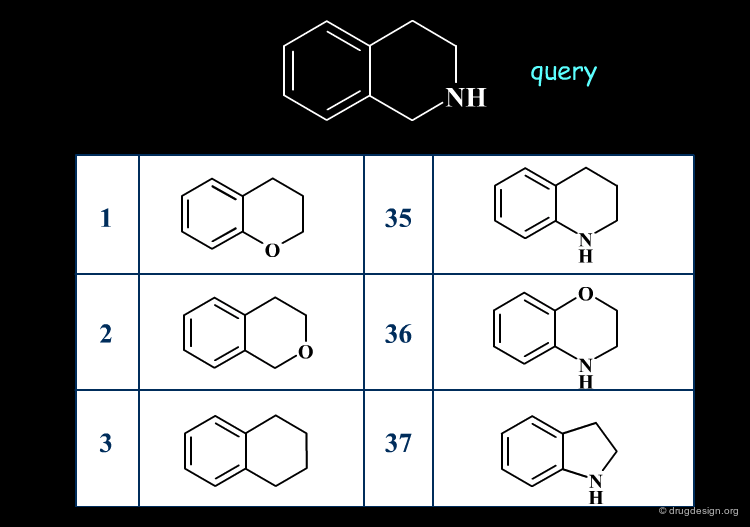
3D Superimpositions¶
A good superimposition in 3D can be the starting point for a new scaffold. For example, the 3D visualization can generate ideas of scaffods based on pharmacophores derived from the alignment of the molecules or from superimposed molecules in the active site of a protein.
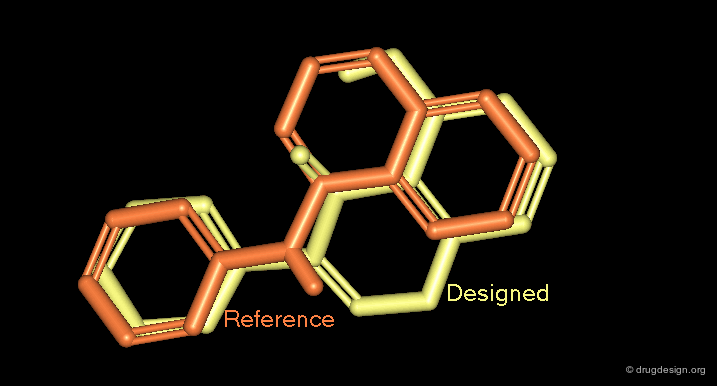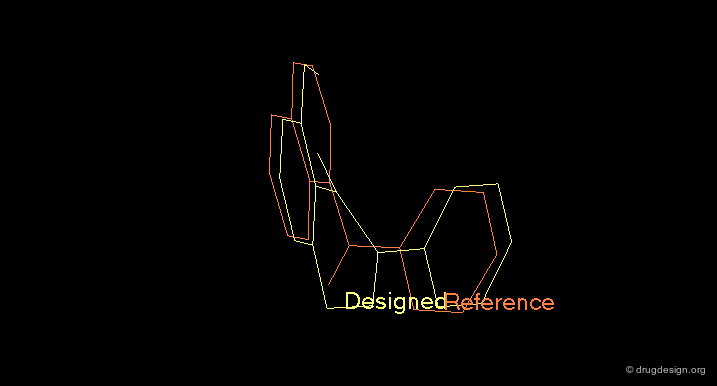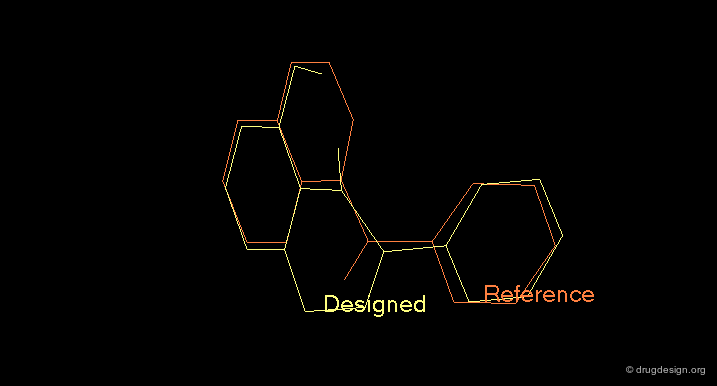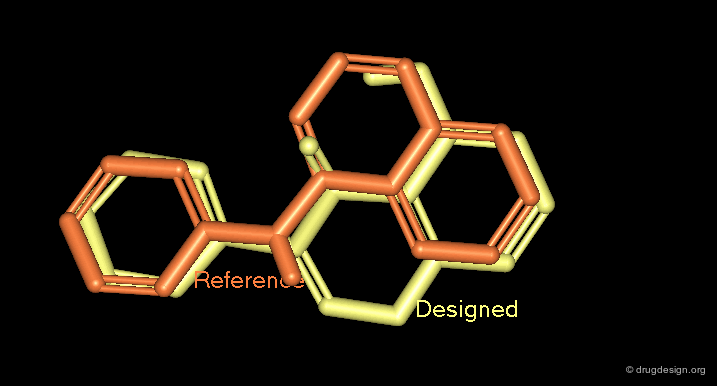
Docking and Virtual Screening¶
The 3D structure of the active site of a target protein can be used to design a new scaffold. For example, based on databases of scaffolds, docking treatments serve to assess the quality of a given structure by first appraising its interactions with the protein, and then considering if the hydrogen atoms of the scaffold are favorably oriented for gaining additional binding with substituents. This can be automated and incorporated in virtual screening treatments.
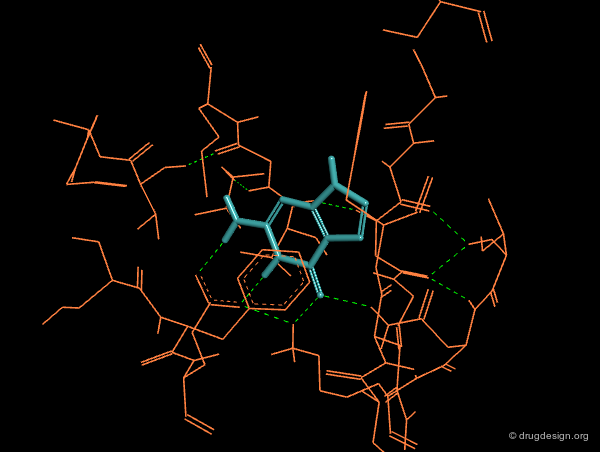

3D Shape Searching¶
Database searching can select molecules that are similar in shape and electrostatic potential to a reference scaffold. A new scaffold can also be identified based on shape complementarity between this scaffold and the protein receptor site.


articles
Shape Signatures: A New Approach to Computer-Aided Ligand and Receptor-Based Drug Design Randy J. Zauhar et al. J. Med. Chem. 46 2003
2D Pharmacophore Searching¶
2D database searching can identify molecules that are structurally diverse with a pharmacophore defined by a 2D-query. In the example below, the pharmacophore derived from an initial cyclic peptide was used as a query for 2D database searching and enabled the discovery of a hit with a much simpler and reduced scaffold. This scaffold was then used for the development of a combinatorial chemistry program.


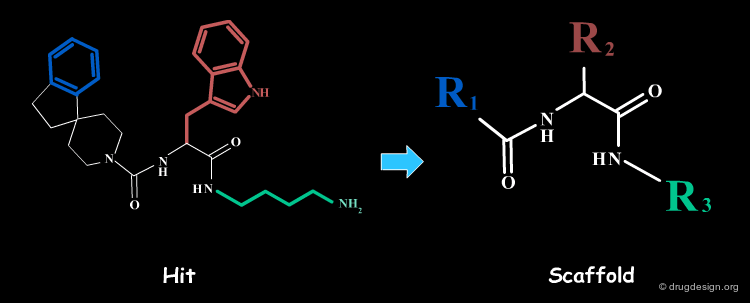
articles
Synthesis and Biological Activities of Potent Peptidomimetics Selective for Somatostatin Receptor Subtype 2 Yang L, Berk SC, Rohrer SP, Mosley RT, Guo L, Underwood DJ, Arison BH, Birzin ET, Hayes EC, Mitra SW, Parmar RM, Cheng K, Wu TJ, Butler BS, Foor F, Pasternak A, Pan Y, Silva M, Freidinger RM, Smith RG, Chapman K, Schaeffer JM and Patchett AA. Proc. Natl. Acad. Sci. USA 95 1998
Rapid Identification of Subtype-Selective Agonists of the Somatostatin Receptor Through Combinatorial Chemistry Rohrer SP, Birzin ET, Mosley RT, Berk SC, Hutchins SM, Shen DM, Xiong Y, Hayes EC, Parmar RM, Foor F, Mitra SW, Degrado SJ, Shu M, Klopp JM, Cai SJ, Blake A, Chan WWS, Pasternak A, Yang L, Patchett AA, Smith RG, Chapman KT and Schaeffer JM. Science 282 1998
Synthesis and Biological Activity of Highly Potent Heptapeptide Analogs of Somatostatin with C-Terminal Modifications Janecka A and Zubrzycka M Endocrine Regulations 35 2001
3D Pharmacophore Searching¶
3D database searching can identify molecules that carry a pharmacophore defined by a 3D-query. This approach provides a means for replacing a reference scaffold by an entirely different one. The method is extensively discussed in different chapters (Pharmacophore-Based Drug Design and 3D Database Searching).

Vector Matching¶
With the idea that a scaffold may just be a spacer connecting the different pieces of a pharmacophore, Bartlett and Cohen developed automated methods to generate scaffolds conforming to the 3D arrangements of a set of substituents. The algorithms rely on 3D vector-based searches. In the example below, an indole moiety proved to be suited for replacing an initial steroid scaffold in positioning correctly the desired substituents.

Hybrids of Known Scaffolds¶
A new scaffold can be designed by examining the superimposition of chemically unrelated molecules and conceived as a hybrid of different molecules. The hybrid scaffold is deliberately different from that of the structures aligned and able to orient properly the various substituents.

Creative Design¶
Creative design results in a combination of several methods. The design summarized below exploited the following: X-ray data revealing the binding of the molecule in yellow (ATP); docking studies of a reference inhibitor (green); 3D superimpositions (that revealed additional overlaps of the ribose and phenyl rings). All this was intelligently exploited to derive an entirely new scaffold (pink), designed through 3D hybridization of the two ligands.
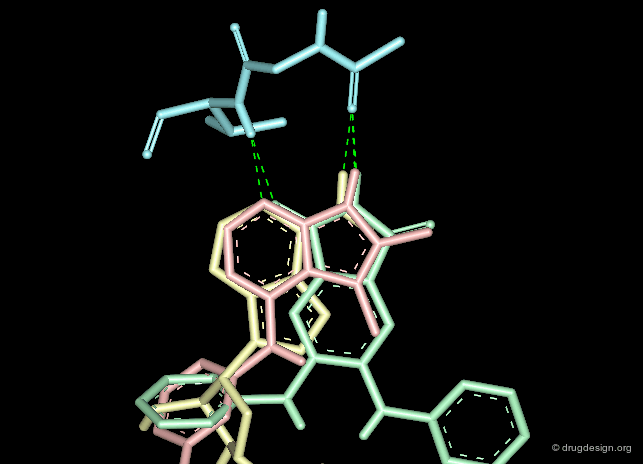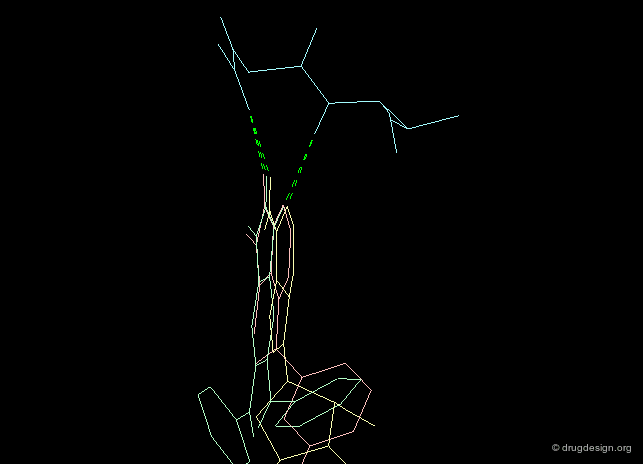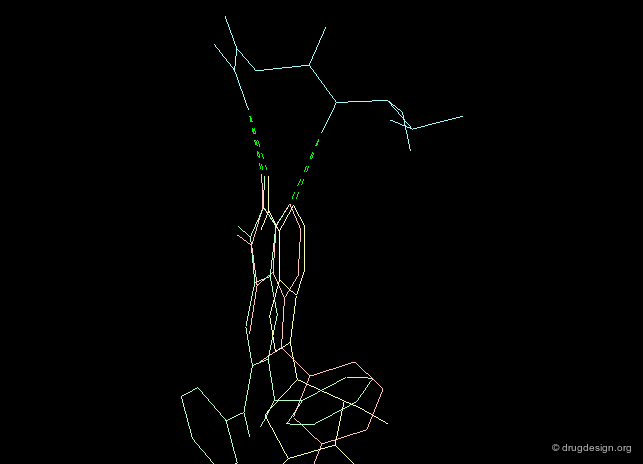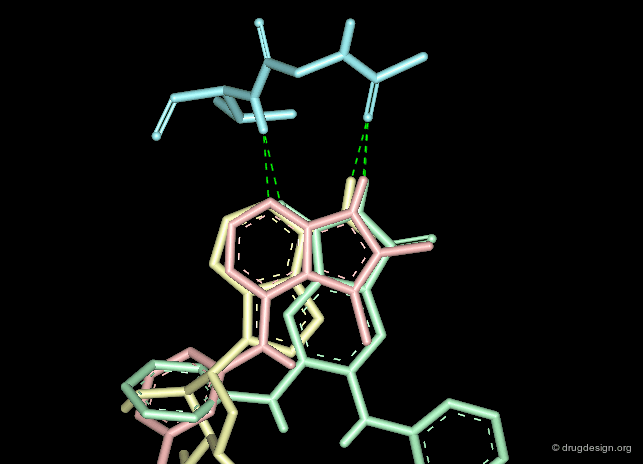
Focused and Diverse Strategies¶
Library Design Goals¶
We will now deal with the strategy for populating the library, and the link between the different molecules. In combinatorial chemistry terminology, this comes down to the nature of the substituents. As will be shown, this is an iterative process that is dynamically conducted with only one aim: to obtain the lead rationally and in a minimum of time. Accelerating the time-to-market translates into priceless advantages.
Library Design Assumptions - Similar Property Principle¶
Library design is based on the "similar property principle" which assumes that structurally similar molecules are likely to have similar properties and structurally dissimilar molecules are likely to have different properties.

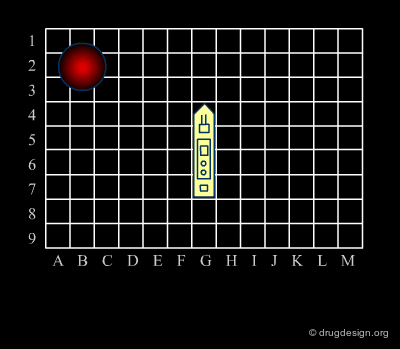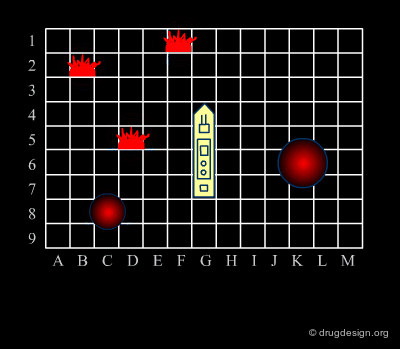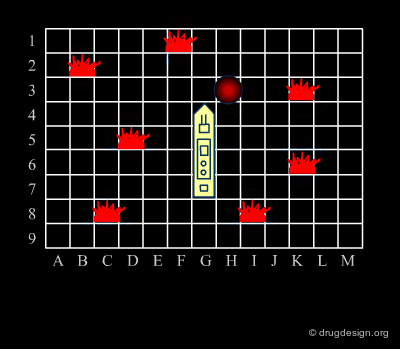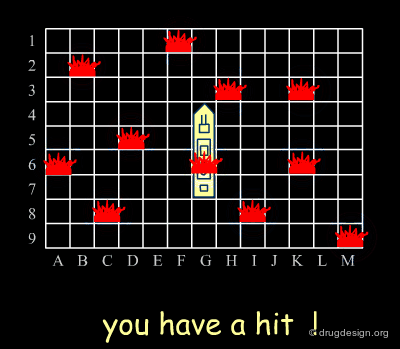
Analogy with Battleships Game¶
Library design can be compared to the popular "Battleship" game, whose strategy develops in two phases. The first is the rapid exploration of the space to identify areas of interest. The second phase consists of a systematic exploration of the area around a hit that has been found. When applied to lead discovery, these two strategies are called "diverse" and "focused" and are presented in the following pages.

Diversity Strategy¶
As in the Battleship game, the diverse strategy (first phase) consists of the rapid exploration of the space to identify areas of interest. Throughout this phase, the keyword is "diversity". The process favors the consideration of diverse chemical structures. This strategy is based on the "similar property principle" already presented and is expected to provide a rapid picture of the molecular space of interest.
Molecular Diversity¶
When applied to drug discovery, diversity is called "Molecular Diversity" and can be defined in two different ways. In the first (structural diversity) the diversity between molecules is expressed in terms of structural considerations, whereas in the second (pharmacophore diversity) diversity is measured in terms of pharmacophore considerations; i.e. the 3D arrangement of elements such as H-bond donors and acceptors, positively charged centers, aromatic rings, hydrophobic centers etc..


Focused Strategies¶
When a specific area appears to be worth exploring, a focused strategy (second phase) is then deployed. In this phase a systematic scanning of the sub-space is deployed meticulously, which allows for the identification of the lead compound that will be the object of further optimization.
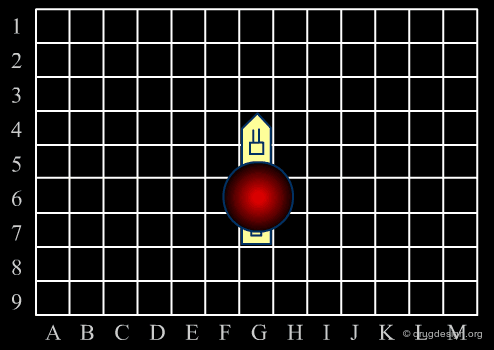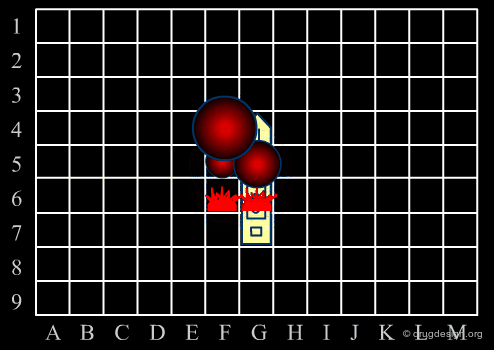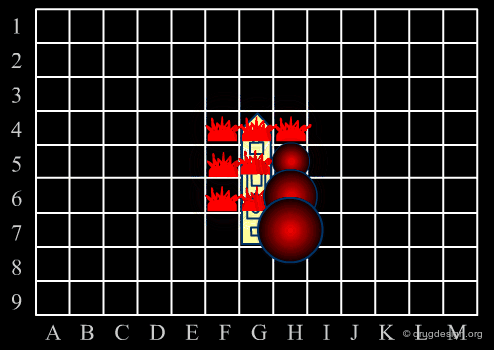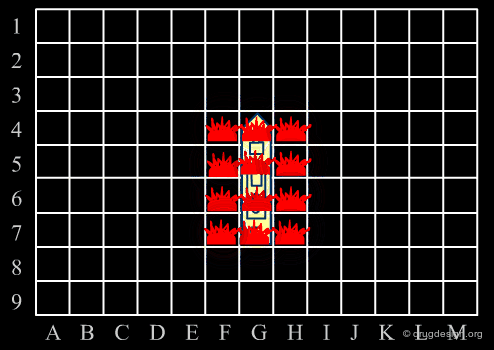
Diverse vs. Focused¶
When applied to drug discovery, diverse and focused strategies should not be opposed: they are neither contradictory nor to be considered as simply appearing one after the other (as in the battleship game). Indeed, they are complementary: "focused" means taking advantage of the information acquired, and "diverse" means avoiding molecules that are useless.

Library Design in the Global Drug Discovery Perspective¶
This graph visualizes how a typical drug discovery project unfolds. With rather limited initial information, a first diverse library allows for the rapid exploration of the space. The strategy consists of the iterative generation and integration of information in the successive rounds (libraries). Throughout this development the libraries become more and more focused. The difficulty is threefold: (1) the extraction of relevant information out of the HTS screening; (2) integration of this information in the design of the next library; (3) finding the right balance between diversity and focused strategies.

Informative Libraries¶
As previously seen, the first priority of library design is to generate knowledge in a convergent manner that enables the identification of a lead. Many HTS screening campaigns generate hits, but no information, and this is a situation with a high probability of going nowhere. Good development of library design should ensure that a maximum amount of information can be obtained while a minimum number of compounds are synthesized and tested (Alexander Tropsha).

Commercial Informative Libraries¶
There are an increasing number of chemical suppliers that propose "informative libraries" also called "targeted libraries" for a variety of targets. In general these libraries are provided on a non-exclusive basis and are based on known scaffolds. Typically these libraries contain fewer than 10,000 molecules and can be purchased and screened, only to produce information. By screening such readily available focused libraries, it is possible to extract predictive structure-activity hypotheses with which to continue to create novel chemically unrelated and proprietary series.


Measuring Distances Between Molecules¶
Methods to Calculate Molecular Similarity¶
In order to quantify the diversity of a given library, we need methods to assess the similarity between two molecules. Some methods will be presented in the following pages. Other words such as similarity, distance or dissimilarity, are also used to designate diversity.
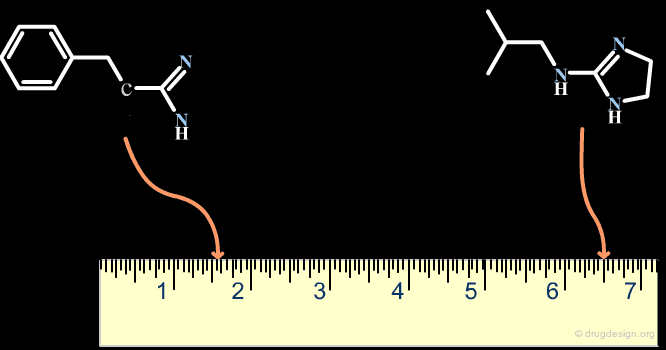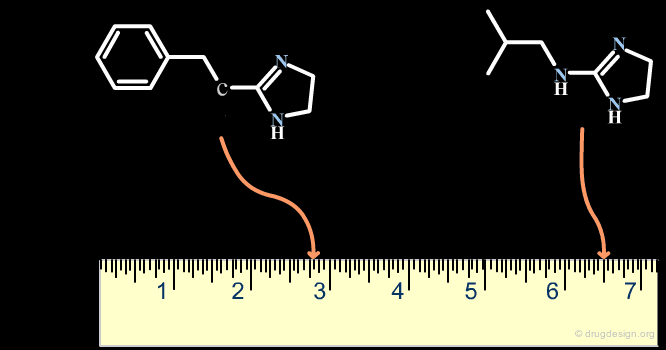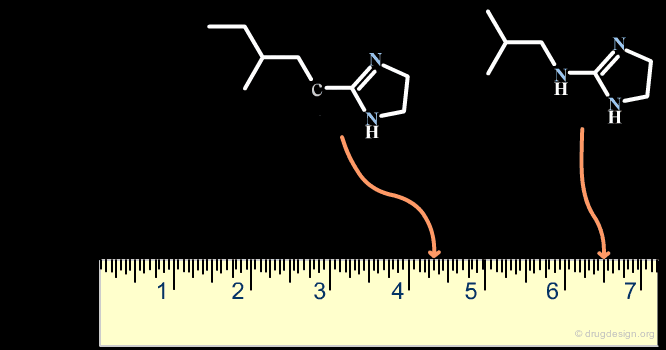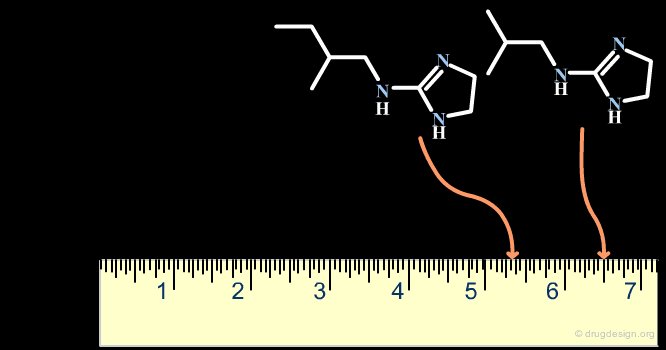
The Distance-Based and the Binary-Based Methods¶
Methods for assessing the degree of similarity between molecules can be divided into two groups. The first includes methods based on the properties of the molecules (called property-based or distance-based methods), and the second includes methods based on their respective chemical structures using binary variables (called structural or binary methods).
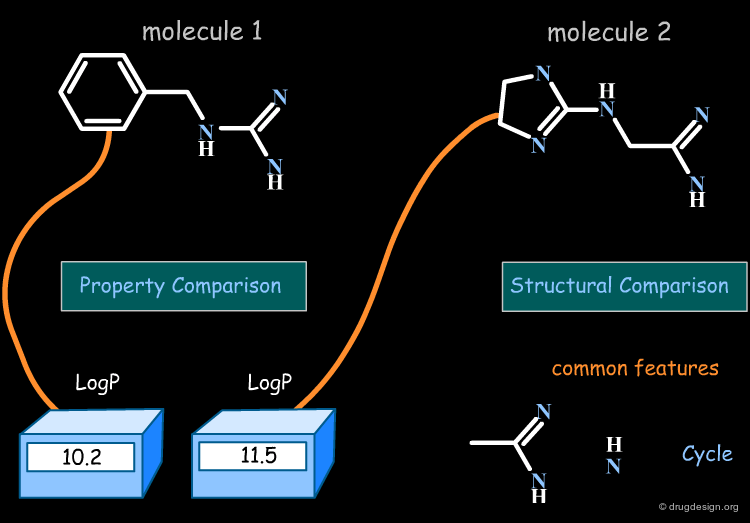
The Property-Based Approach¶
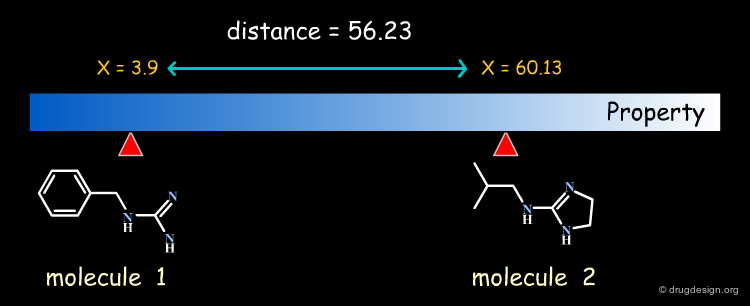
In property-based methods the "distance" between two molecules is calculated directly, based on the numerical values of the property for each of the two molecules compared.
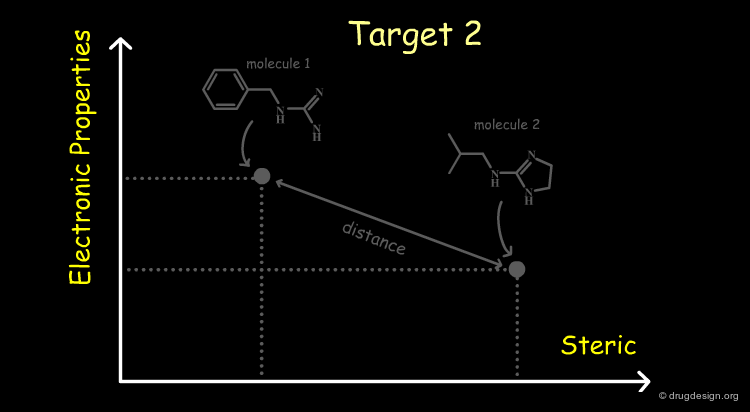
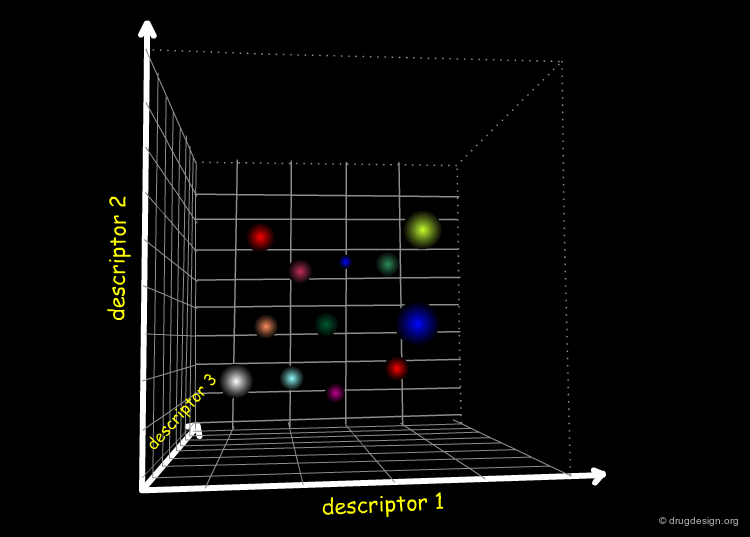
Molecules in the Space of their Relevant Properties¶
Relevant properties should be considered for calculating the "distance" between two molecules. In the following diagram the molecules are represented in the space of their properties, each axis representing one relevant property. It is also important to note that the relevant properties depend on the nature of the target (properties for target 1 are different from those for target 2).


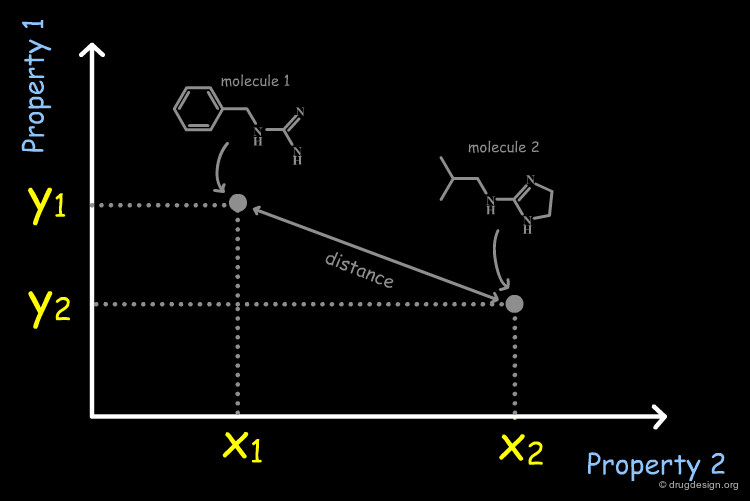
From Molecular Properties to Molecular Descriptors¶
It is necessary to quantify each of the individual properties (x1,x2,y1,y2 values) in order to derive a dissimilarity measure. However it is often difficult to calculate them directly from the molecular structure. In current practice, it is easier to replace the molecular properties by structural factors (known as molecular descriptors) related to the molecular property concerned, which can be simply calculated.
High-Dimensionality Space of the Molecular Descriptors¶
Since a property is not represented by one sole descriptor and since many properties are involved in a biological system, the "chemical diversity space" will be characterized by a high degree of dimensionality in the referential of the molecular descriptors.
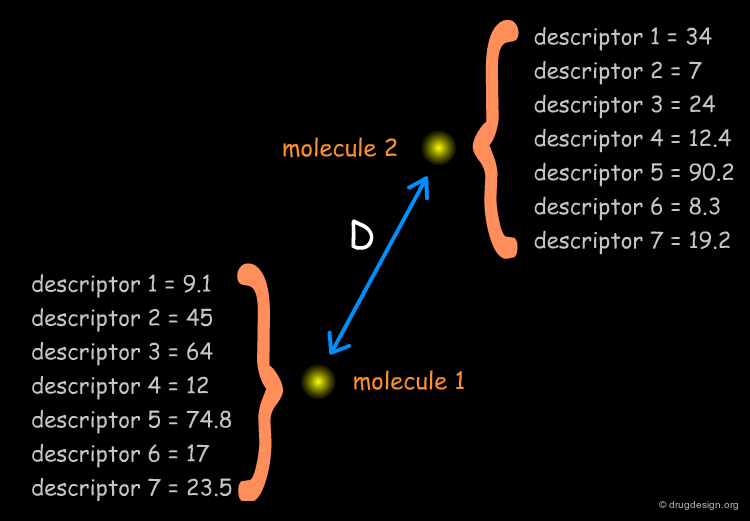
Similarity Coefficient and Distance Coefficient¶
As we identified the descriptors and calculated them for each molecule, let us now address the mathematical issues involved in measuring the diversity between two molecules. Some methods calculate the absolute "distance" directly (the "distance coefficient D"), whereas others generate normalized results (the "similarity coefficient S"), as a measure of the diversity. The Tversky, Tanimoto, Euclidean, Hamming and cosine measures are the most frequently used equations, and some of them will be described in the following pages.
Euclidian Distance¶
The Euclidian distance equation is straightforward and provides the distance (or dissimilarity) between pairs of molecules.

Tanimoto Coefficient¶
The Tanimoto coefficient is derived from the Tanimoto equation, one of the most popular ones for measuring similarity. It corresponds to a direct measure that includes a size normalization of the denominator term, therefore the results are not biased by the size of the molecules.

The Structural Approach¶
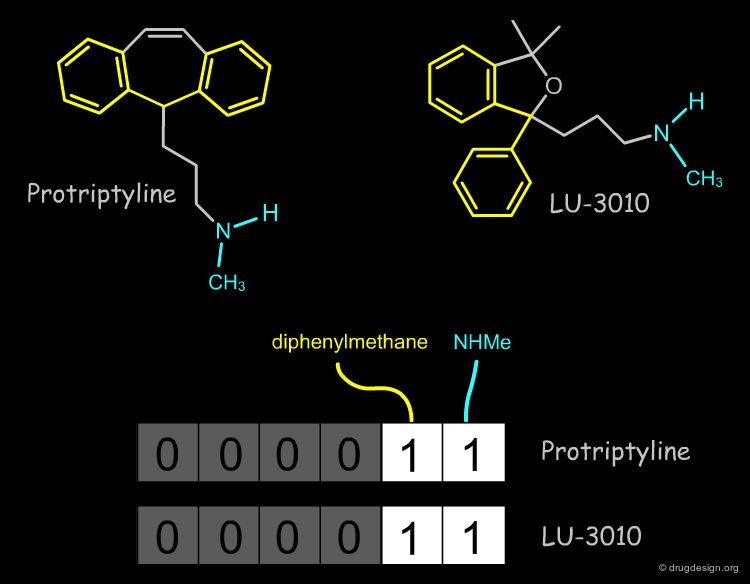
The structural approach is based on bitmaps (often referred to as "fingerprints") and have bits set according to the presence or absence of certain structural features in the molecule. Each bit represents a structural descriptor. The premise of the similarity comparison is that similar structures have similar fingerprints. An example of a simple fingerprint is shown below.

Relevant Structural Keys¶
To make a fingerprint, one decides which structural features (patterns) are relevant and assigns a bit to each of them.
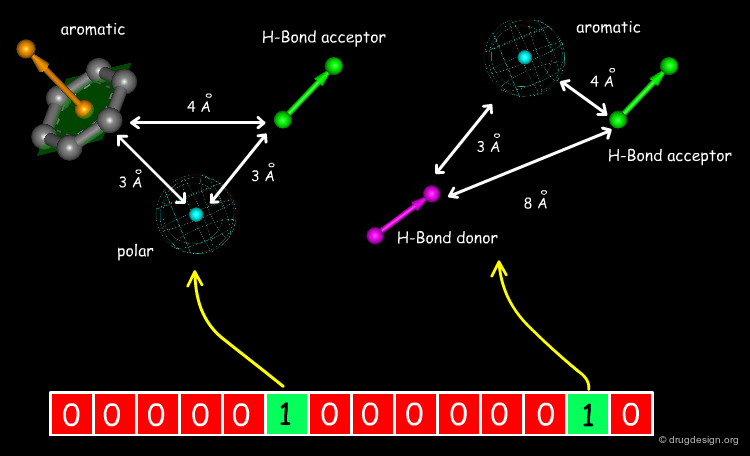
Extended 3D Fingerprints¶
Initially developed for 2D chemical structures, the concept of fingerprint has been extended to 3D pharmacophores. In this approach pharmacophores are reduced to 3-4 points with their corresponding distances (these points include H-bond donors, H-bond acceptors, hydrophobic centers etc...). Each pharmacophore arrangement is represented in the fingerprint by a bit position.
Example of Structural Key¶
An example of a fingerprint is represented below. Moving the cursor on a given bit (0 or 1) in the diagram, allows you to identify the particular structural feature concerned.

Similarity Coefficients and Distance Coefficients¶
Now that we have generated the fingerprints for all the members of a database, let us address the mathematical issues for measuring the diversity between two molecules. The same methods presented in the property-based approach (Tanimoto, Euclidian, Hamming, cosine etc...) can be applied with formulas adapted to binary variables.

Binary Tanimoto Example¶
The meaning of the Tanimoto coefficient for measuring the 2D structural diversity of two molecules is illustrated below. A and B are the number of bits set in the fingerprints of molecule A and B, respectively; C is the number of bits that are set in both molecules. Very similar molecules have a Tanimoto structural index greater than 0.8 (80%)

Computational Speed¶
Generating the structural keys for an entire database is time-consuming. However once the fingerprints have been generated, the calculations become extremely rapid (pair wise comparison of millions of bits can be made by a computer in a millisecond).

Similarity Index of an Entire Library¶
We now know how to calculate the similarity between two molecules. It is also possible to calculate the similarity index of an entire library. The sum of the distances between all possible pairs of molecules present in a library, divided by the total number of molecules, can give an indication of the diversity of this library. Designing a focused library means keeping this index small, and for diversity it should be large.
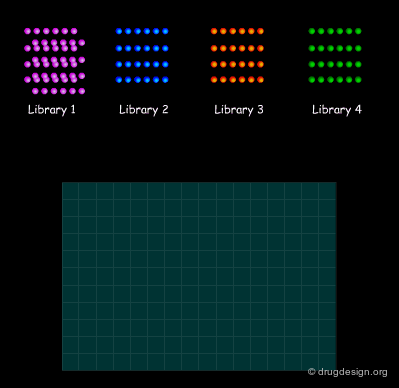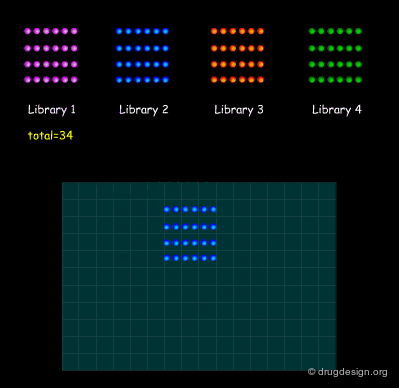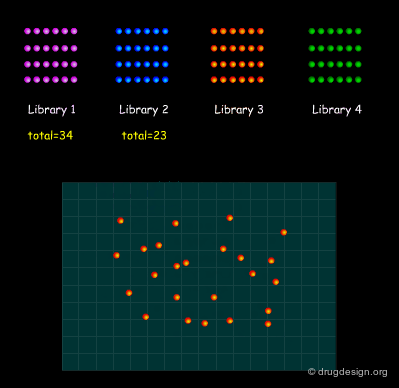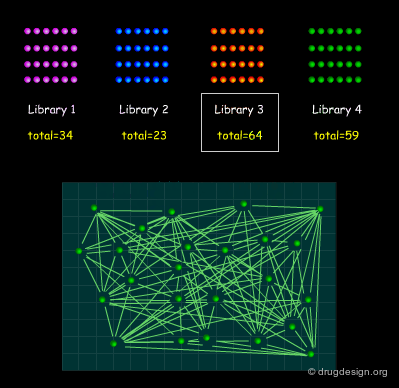
Huge Dataset of Undigested Information¶
Ideally, one should keep the dimensionality of the space relatively small, however this is not always possible (for example, overly limited initial data do not allow for the extraction of a small number of relevant descriptors). If we have 150 structural keys or 35 molecular descriptors, we are confronted with an ocean of undigested information, where it is impossible to analyze or visualize any trend whatsoever!
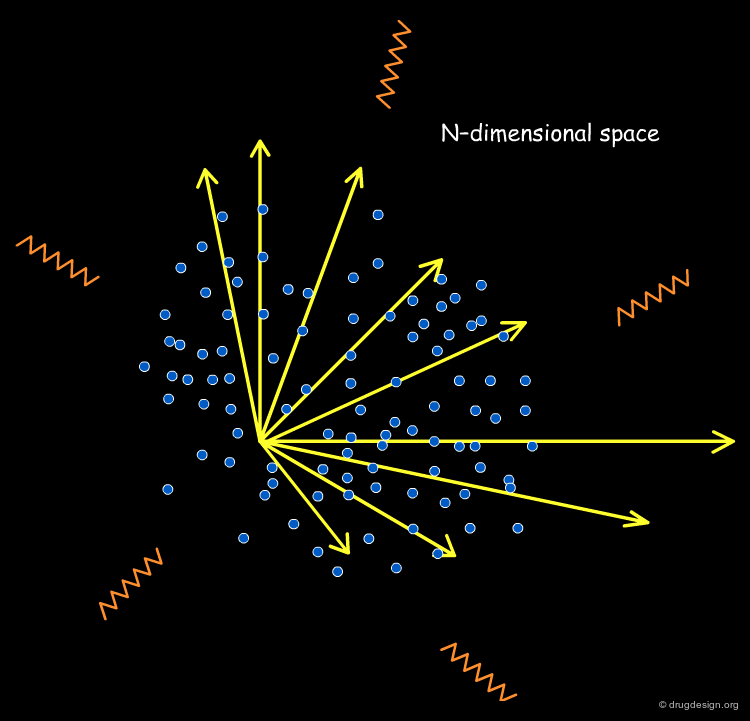
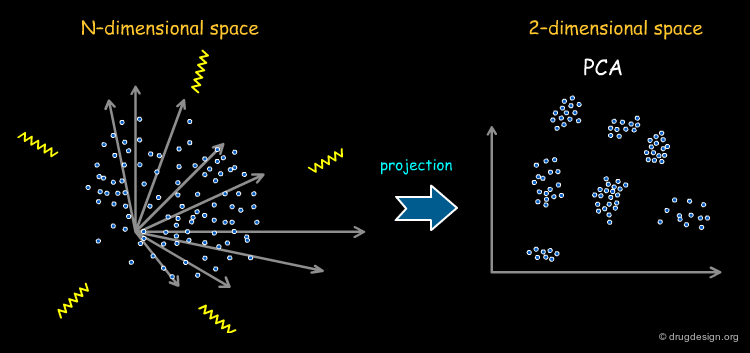
Principal Components Analysis¶
Principal component analysis (PCA) is a technique to simplify a dataset and to reduce the problem dimensionality by retaining those characteristics of the dataset that contribute most to its variance. It is therefore convenient to reduce the dimensionality of the full set of descriptors by PCA before they are used in diversity analysis. This representation also enables the models to be visualized easily. The weakness of this method is the difficulty of interpreting the results in structural terms (the content and meaning of the axes in PCA is far from being straightforward).
Subset Selection Issues¶
Subset Selection Problem¶
In the last section we learned how to find relevant descriptors, and how to calculate the "distances" between molecules to score libraries. As the virtual library includes a huge number of compounds, what remains to be found is a method for reducing it to a practical size to be synthesized. This is known as the "subset selection problem".

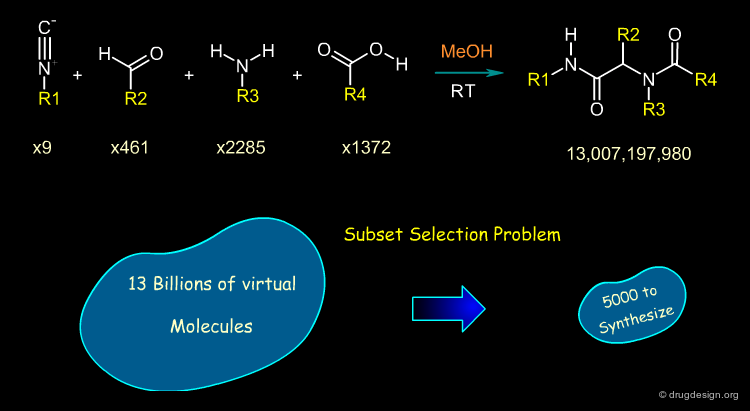
Illustration of the Subset Selection Problem¶
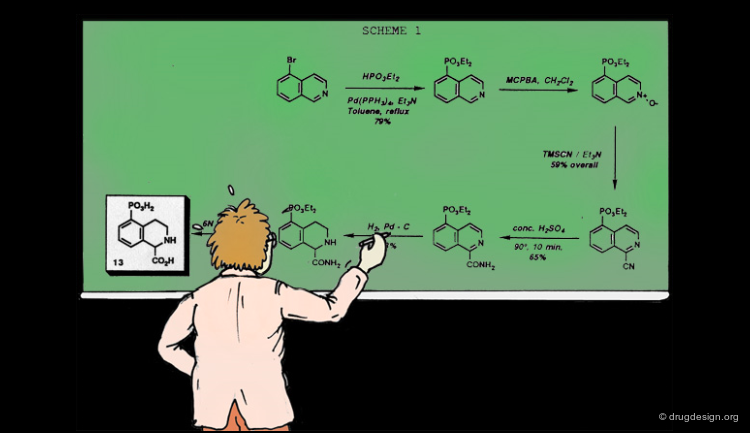
To illustrate the "subset selection problem" we see below the combinatorial scheme of a four-component Ugi reaction. This scheme defines 13 billions of molecules and we need to reduce them to a more reasonable number of molecules to be synthesized!
The Systematic Route¶
In theory we can generate all possible subsets, calculate the diversity of each one, and the most diverse subset (with the highest diversity value) will be adopted for the combinatorial chemistry.

Systematic Assessment Impracticable¶
Exploring all possible subsets of size p within a pool of N molecules is not computationally feasible for typical values encountered in library design. The program implemented below allows you to calculate the number of possible subsets of size p from a dataset with a total of N molecules.

Solving the Subset Selection Problem¶
There is no exact solution to the subset selection problem and approximate methods are needed to make the library design useful for practical situations. The most common methods for compound selection that have been developed fall into three categories: distance-based, clustering and optimization methods.

articles
Analysis of selection methodologies for combinatorial library design Rosalia P. et al. Molecular Diversity 6 2003
Distance-Based Methods¶
Distance-Based Methods (also called dissimilarity-based) aim at identifying a diverse set of molecules directly using for example, Euclidian or Tanimoto metrics. An example of implementation is illustrated here. The process starts by selecting a compound at random and identifying the molecule in the database that is most dissimilar. That molecule is then added to the subset of the initial compound and the process is repeated until the subset is of the desired size.

Clustering Methods¶
Clustering is a procedure in which objects (molecules) are divided into groups. In library design each compound is associated to a cluster so that compounds in the same cluster are closer to one another (in a distance space involving all the property values). The clustering process is computationally expensive, whereas the subsequent molecule selection is very trivial. Diversity is achieved by selecting a compound from each cluster.


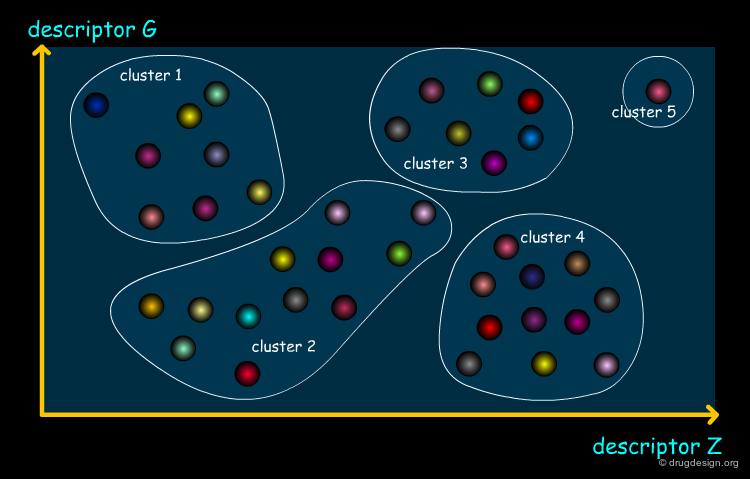

Cell-Based Partitioning¶
Cell-based partitioning is a particular clustering method involving a dimension reduction of the chemical space. Each property is divided into "bins" whose combinatorial product defines a set of cells that cover the entire space. Each compound is assigned to a cell and a subset can then be chosen by selecting one molecule from each cell. An attractive feature of this method is that it does not require the calculation of distances between molecules.

Optimization of Diversity Function¶
This method is based on a "diversity function" that is to be optimized. An initial set of molecules is chosen and further improved by successive iterations, until convergence is achieved. The diversity function has been the object of intense development interest, the most common functions currently used are MaxMin and MaxSum.
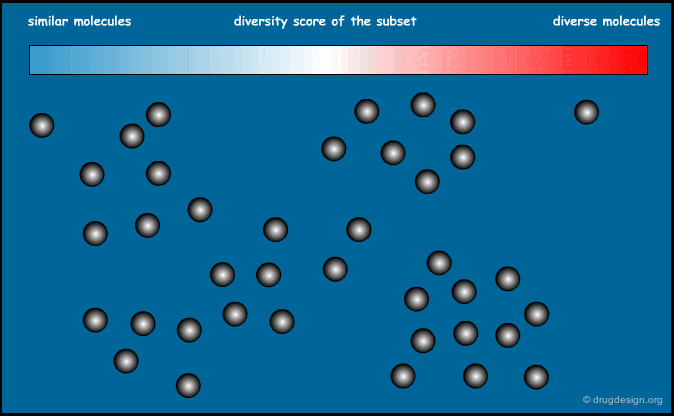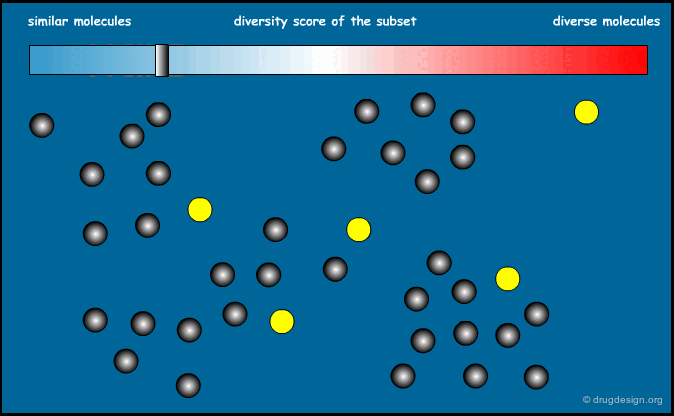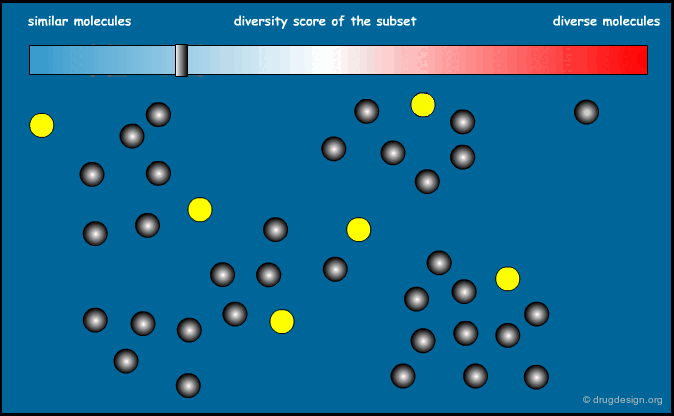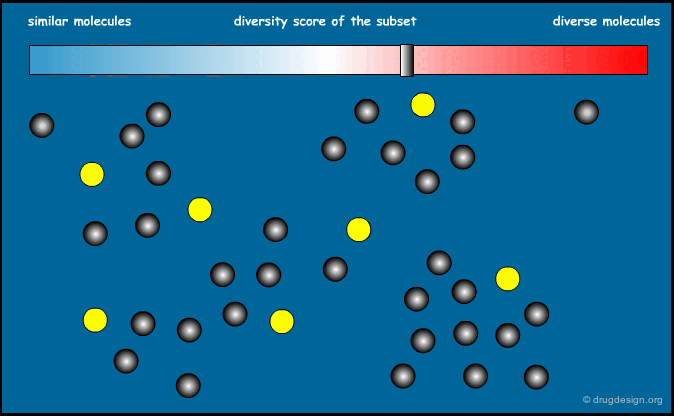
Principle of the Optimization¶
The principle of the optimization is based on the use of a function (the "diversity function") to be optimized, which allows deciding where to go. This is achieved by maximizing diversity (or similarity), iteratively, by means of a stochastic algorithm such as simulated annealing, Monte Carlo, genetic algorithms etc...

Example of Selection of Diverse Compounds¶
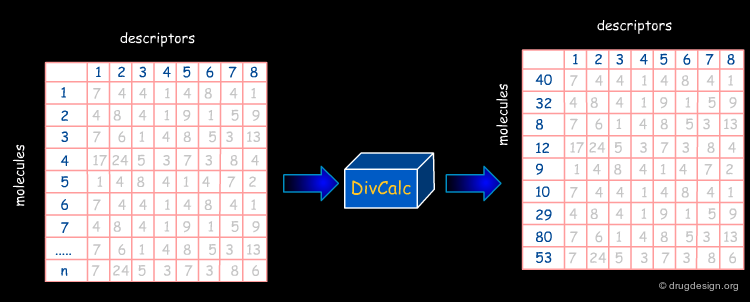
In the following pages we will present the use of DivCalC, a program addressing the selection of diverse subsets from larger collections of chemical compounds. Based on the combination of molecular descriptors and appropriate scaling, the program is used to select a given number of the most diverse compounds. DivCalc uses distance based dissimilarity algorithms.
articles
DISSIM: A program for the analysis of chemical diversity. Flower, D. J. Mol. Graphics Mod. 16 1998
The Input¶
An example of input data is shown below in an Excel-like grid. The rows represent molecules and the columns represent descriptors.

Normalization of the Data¶
Sometimes the descriptors calculated have very different ranges in which they can occur and the Euclidean distance can be unnecessarily biased by non-normalized values. To avoid this, data normalization provides a common scale for all the molecular properties. The data are scaled by using the mean and standard deviation for each property, as illustrated below.

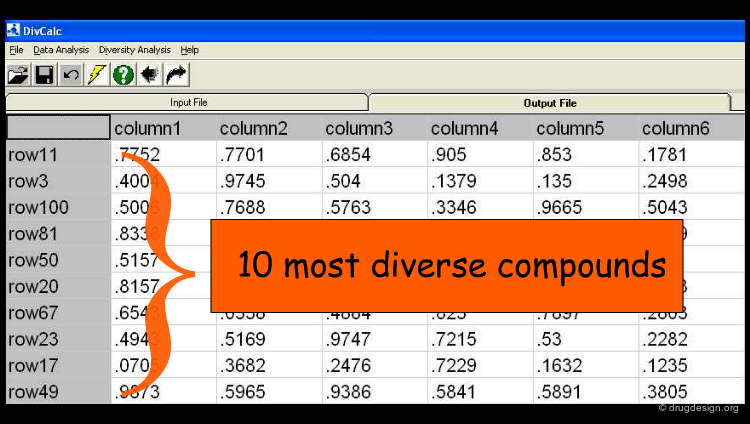
The Results¶
The user can set a predefined limit on the number or percentage of compounds to be displayed in the output (e.g. the 10 most diverse compounds). By default, all rows (compounds) in the input are ranked by their diversity and are shown in the output.
Reagent Selection¶
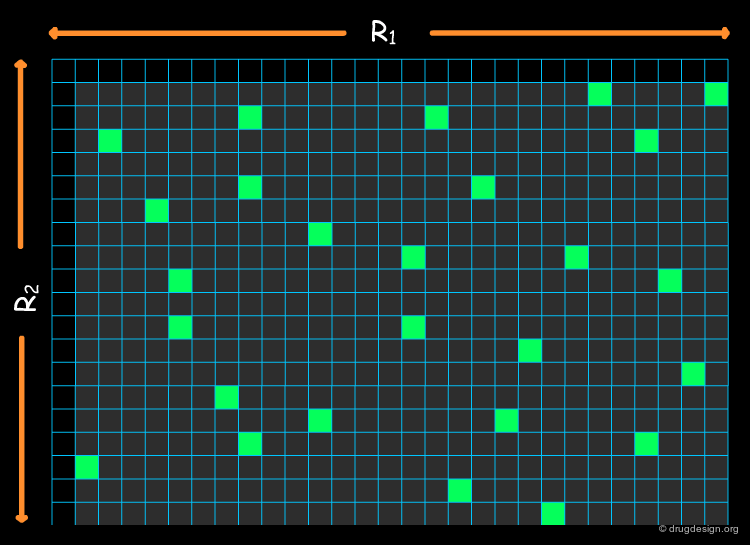
Cherry Picking Limitations¶
In the previous section we discussed some subset selection solutions. When only diversity is the goal, the approach is called "cherry picking". This is only useful for the purchase of novel compounds but not for the development of a combinatorial chemistry program: it is synthetically inefficient because the combinatorial constraint is not taken into account.
Optimization of Diversity and Synthetic Issues¶
To be practical, it is necessary to find a compromise that satisfies both the diversity requirements and the combinatorial chemistry constraints.
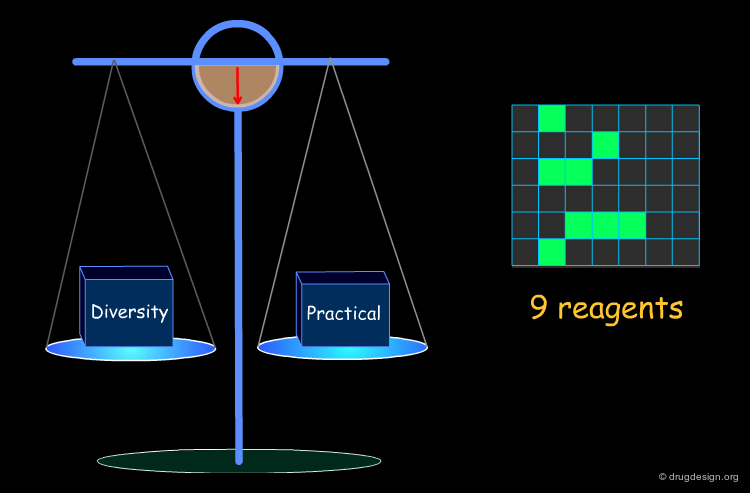
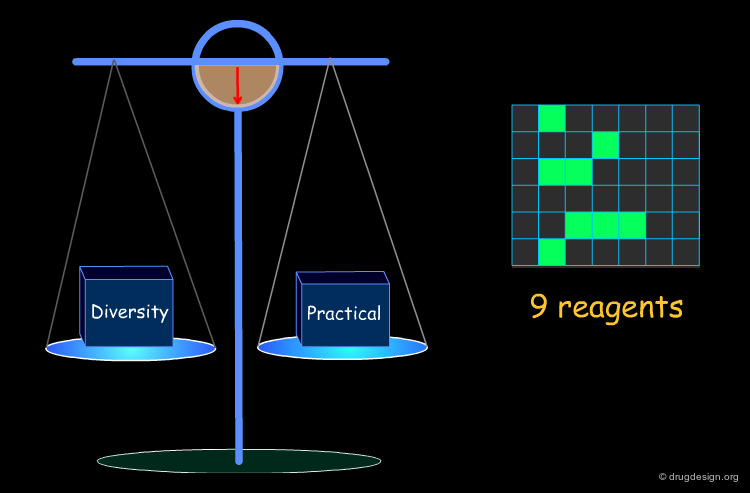


Example of Optimization Algorithm¶
A simple example of an optimization algorithm is illustrated below. The idea is to direct diversity (only one reagent is added at a time). Most important is that at each step the list of already selected reagents is optimally exploited, so that the total number of reagents is continuously kept to the minimum.
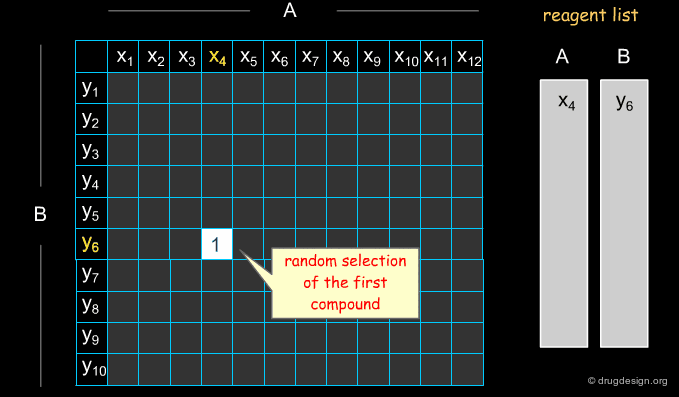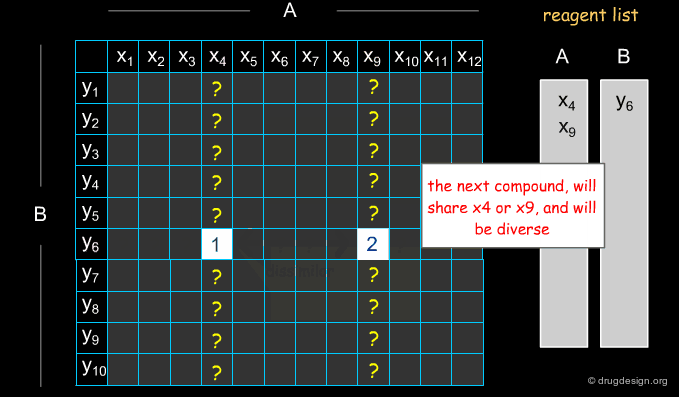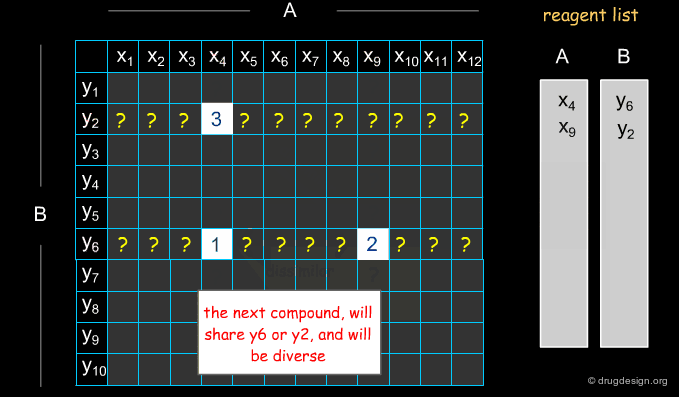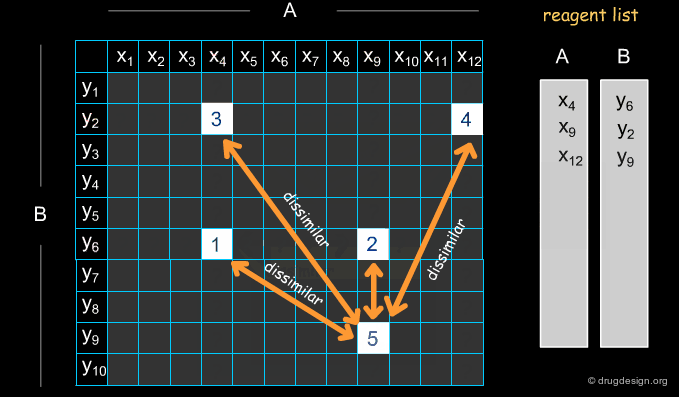
Selecting Reagents is a Complex Issue¶
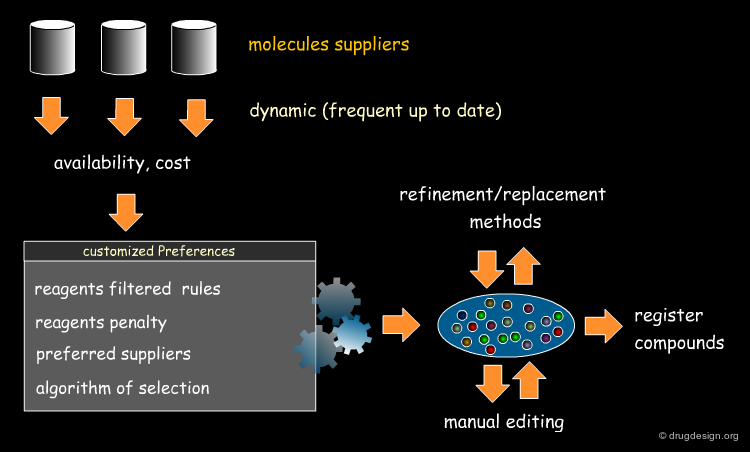
Selecting reagents is a complex multi-dimensional optimization problem. Many parameters need to be considered in an integrated manner. The list of available reagents needs to be constantly kept up to date; weights are assigned (e.g. to each reagent or supplier) that increase their chances of being selected in the optimization process; delivery time need to be integrated; the system must allow for editing, removing, changing, replacing etc... automatically or manually, at any time.
Increasing the Quality of a Library (ADMET)¶
Failures in Clinical Trials¶
Statistical analyses indicate that more than 50% of drug candidates that enter into clinical trials are discarded because of inadequate pharmacokinetics (ADME) and toxicity properties. In addition, some drugs are taken off the market after successful clinical trials. Of the $800 million needed to get a new drug on the market, it is estimated that more than $400 million are wasted in pursuing unproductive leads.

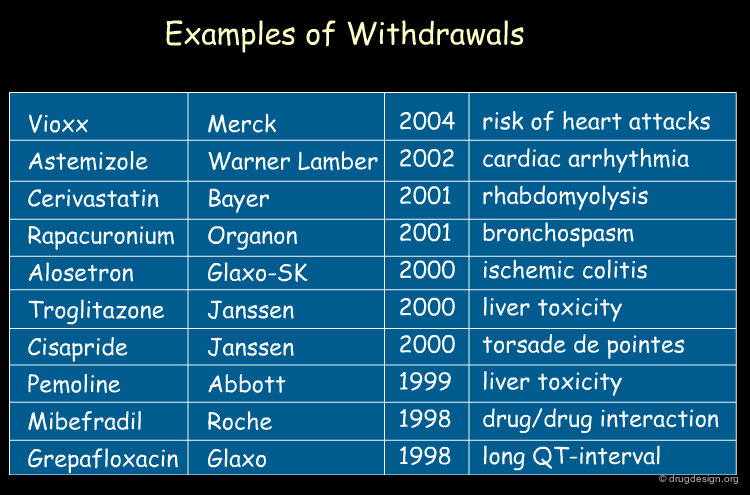
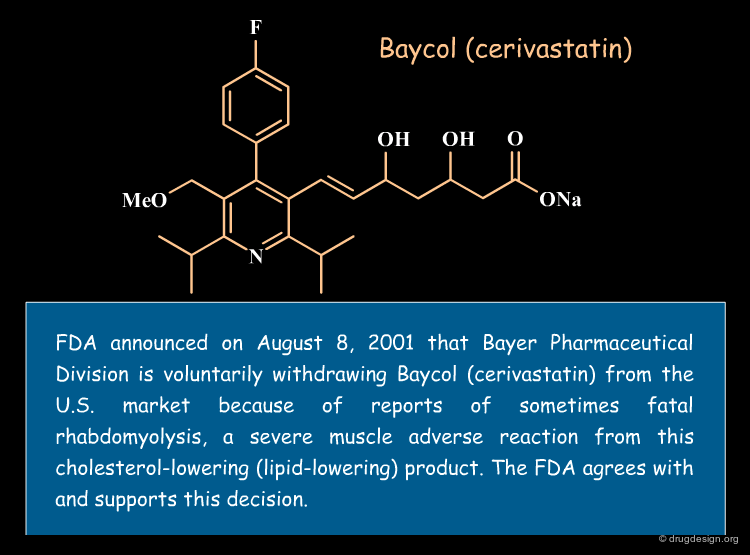
articles
Managing the Drug Discovery/Development Interface Kennedy T Drug Discov. Today 2 1997
Failure in Drug Discovery¶
Historically, this poor productivity can be explained by the impracticality for the medicinal chemist to develop a global approach to drug discovery. Focusing on potency and selectivity, other considerations such as solubility, bioavailability, ADME properties etc... were beyond his concern, and also beyond his expertise. A lead was often selected with insufficient assessment of its drugability and with a structure that did not always permit sufficient optimization of all the properties required.

Early Integration of ADME Properties in Drug Discovery¶
Today, with the progress in combinatorial chemistry and HTS, finding a hit is no longer a problem. The central issue becomes the identification of a hit that will direct to the best lead, prior to a costly optimization effort. To do so, the medicinal chemist needs a more global approach that allows him to identify dead-end leads and also to integrate ADME considerations early in the drug discovery process.

The Drug-Like Approach and the Predictive Approach¶
The first attempts at early integration of ADME properties in drug discovery, started to appear in the nineties and were guided by the "drug-like" principle. More recently, a predictive "computational approach" has also emerged. The two approaches will be presented in the following pages.

The Drug-Like Approach: Identify Poor Candidates¶
In the "drug-like" approach, to increase the chances of a lead becoming a drug, its structure should resemble that of a known drug (that has proven ADME properties). A lead with no such resemblance will therefore introduce an unnecessary risk. The aim of this approach is therefore to eliminate compounds with poor ADME characteristics early in the discovery process. It has been estimated that by doing so drug companies could save up to 60 million dollars per drug.
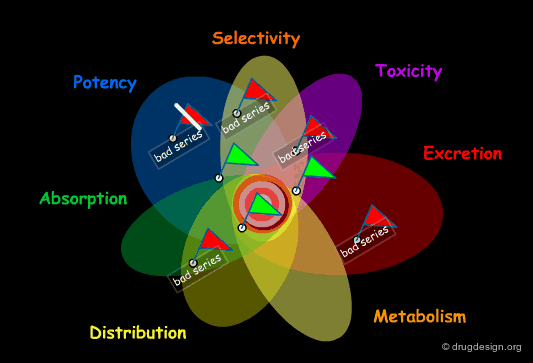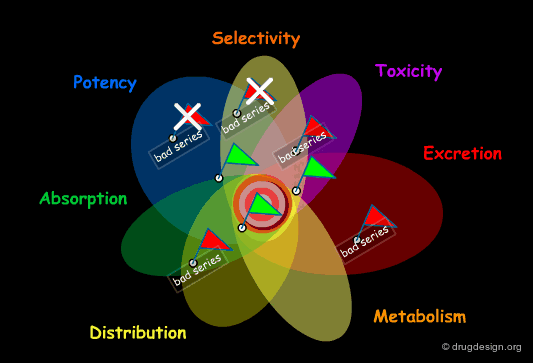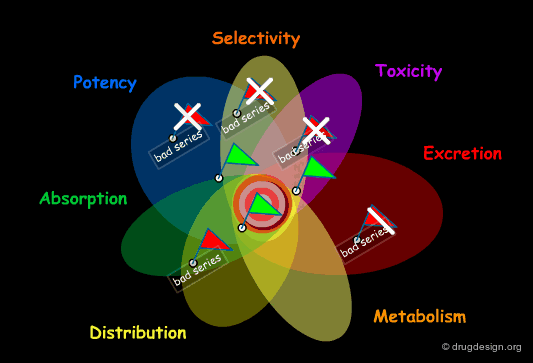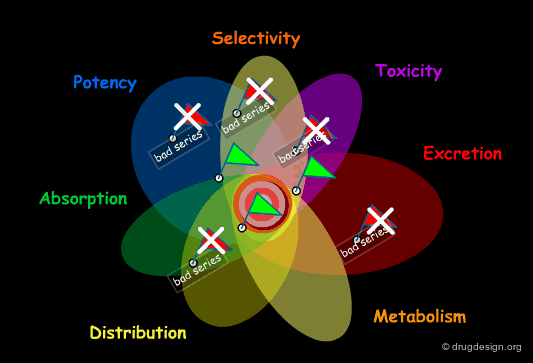
Structural Mimicry and ADME Properties Mimicry¶
In the "drug-like" approach the resemblance is either based on the structure of a reference drug (in the example below: a benzodiazepine scaffold), or on the resemblance of the various important physico-chemical parameters of that reference drug (ADME properties mimicry).


The Rule-Based Approach¶
In order to provide guidelines for mimicking favorable ADME properties observed in known drugs, extensive studies were made to analyze the numerous parameters that influence these properties directly or indirectly. For example drug databases such as the World Drug Index (WDI) that contain information on more than 70,000 pharmacologically active compounds, including all marketed drugs, served as a basis for deriving empirical rules to filter and eliminate the most obviously poor candidates. Lipinski was one of the first pioneers in this area.

Lipinski Rules (Rule of 5)¶
By analyzing the properties of molecules in the World Drug Index, Lipinski developed the now popular "Lipinski's rule of five" (named because of its emphasis on the number 5 and multiples of 5) which predicts in a simple manner the drug-likeness of a molecule. Note that these rules aim at the elimination of poor candidates, and do not assess whether a given compound is good.

articles
Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings C.A. Lipinski et Al. Advanced Drug Delivery Reviews 23 1996
MW Distribution¶
The following diagram illustrates the profile of a library in terms of the molecular weight distribution. It shows that most compounds comply well with the Lipinski rule associated to the molecular weight (MW less than 500).

LogP Distribution¶
The following diagram illustrates the profile of a library in terms of the LogP distribution. It shows that most of the compounds comply well with the Lipinski rule associated to LogP (LogP less than 5).

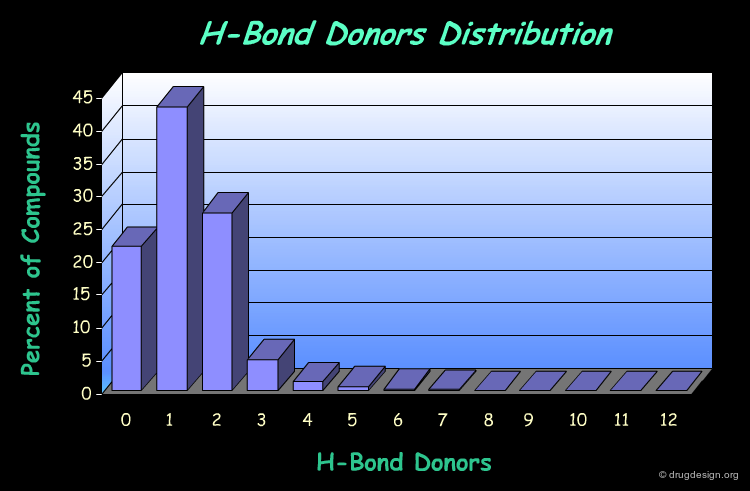
H-Bond Donor Distribution¶
The following diagram illustrates the profile of a library in terms of the H-bond donor distribution. It shows that most of the compounds comply well with the Lipinski rule associated to this property (less than 5 H-bond donors).
H-Bond Acceptor Distribution¶
The following diagram illustrates the profile of a library in terms of the H-bond acceptor distribution. It shows that most of the compounds conform well to the Lipinski rule associated to this property (less than 10 H-bond acceptors).

Total Analysis¶
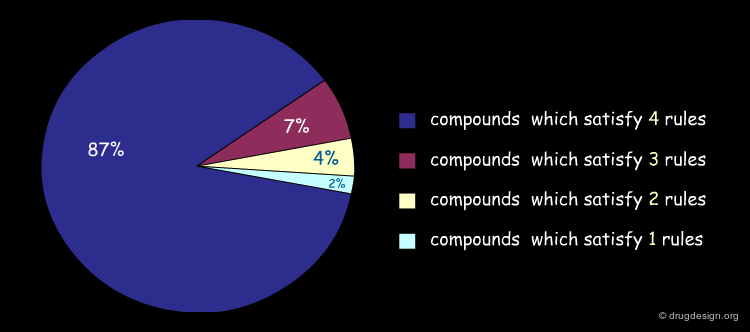
It is possible to represent visually the extent to which the compounds of a library comply with the Lipinski rules. Below is illustrated an example of analysis of the content of a library, and the percentage of compounds that satisfy several rules simultaneously. Note that molecules which only satisfy 3 rules are not always eliminated (this depends on the problem to be solved).
Other Rules¶
Following the idea initiated by Lipinski, other rules were developed and are currently used in the primary filtering of molecules for high through-put screening. These rules are not always related to ADME properties some of them were introduced to get rid of molecules which were difficult to optimize (e.g. many chiral centers etc...). The common characteristics of all these rules is that they can be calculated quickly and allow for the real time filtering of millions of compounds.

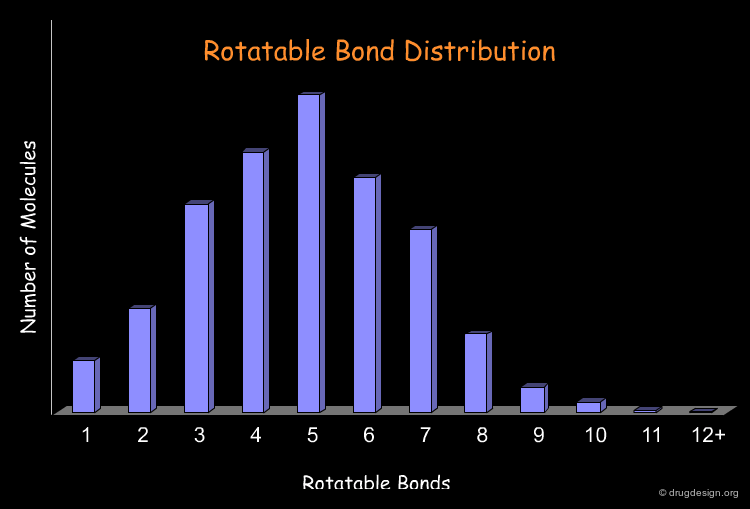
Remove Too Flexible Molecules¶
The number of rotatable bonds is often used as a filter to identify possible drug-like molecules. Retrospective analyses show that 70% of the drug-like molecules have between 2 and 8 rotatable bonds. There is a general consensus that overly flexible molecules must be rejected (e.g. with more than 12 rotatable bonds). The following diagram illustrates the profile of the molecules of a library in terms of the rotatable bond distribution.
Remove Molecules with too Many Rings¶
Analyses reveal that 70% of the drug-like molecules have between 1 and 4 rings. The filtering process will therefore eliminate molecules with more than 4 rings. Sometimes the size of the rings are also considered: molecules with rings higher than seven-membered are eliminated.
Remove Compounds with Known Toxic Moieties¶
It is not rare to observe that drug candidates exhibit detrimental toxicity in late development. Toxicology has accumulated experience and collected data enabling the identification of "toxicophores" (see below). These are structural patterns that have been observed to be associated with toxicities.

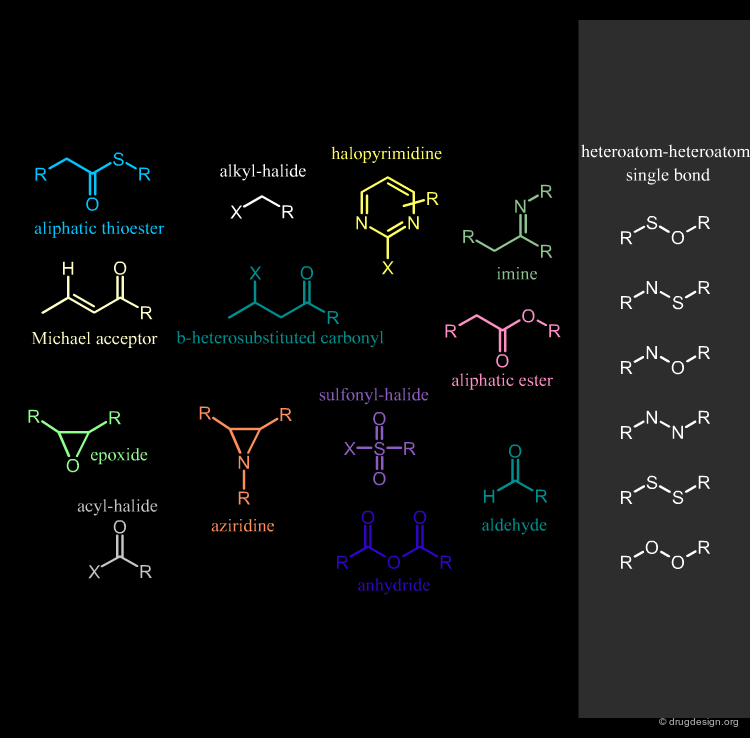
Remove Compounds with Reactive Groups¶
Compounds that contain chemically reactive groups are likely to be toxic, unstable or may interfere with the biological assays and also lead to false positive results. This filter is one of the most debatable since drugs with reactive functions exist. Examples of chemically reactive groups are shown below.
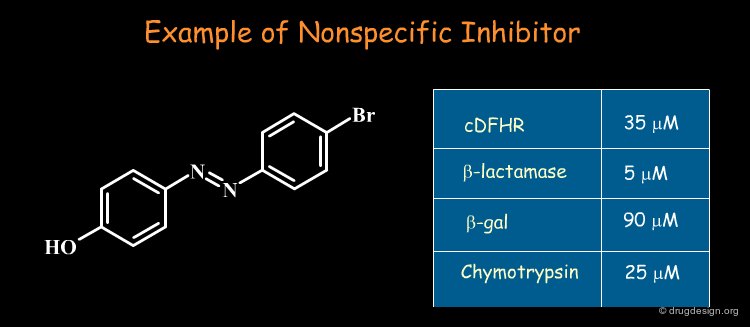
Remove False-Positive Hits¶
Some molecules emerge repeatedly as hits in diverse unrelated target systems. These so-called "promiscuous" hits act non-competitively, show little structure-activity relationships, and have poor selectivity: they artificially inflate hit rate in screening. Their mechanism of action remains unknown (in some cases aggregates are formed that are the inhibitory species). These types of non-specific molecules should be recognized and removed from any biological assays!
articles
A Common Mechanism Underlying Promiscuous Inhibitors from Virtual and High-Throughput Screening - aggregation Susan L. McGovern J. Med. Chem. 45 2002
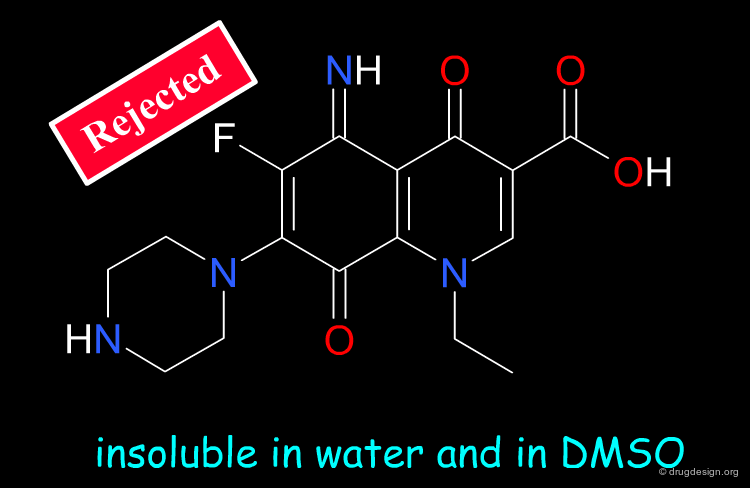
Remove Poorly Soluble Compounds¶
Molecules that have poor solubility create problems not only for the primary screening but also for further development. It is general practice that compounds that are likely to be insoluble in DMSO/Water (10%) should be eliminated.
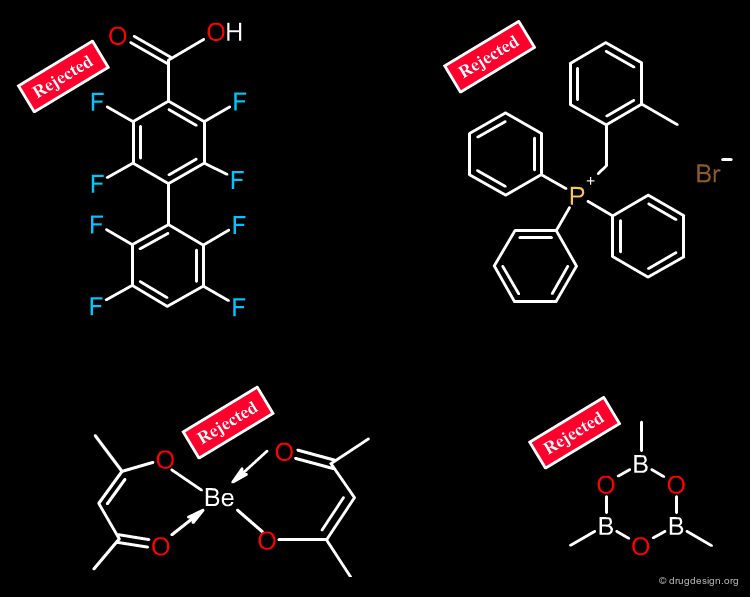
Filter on Heteroatoms and Non-Organic Molecules¶
Molecules that have too many halogen atoms (e.g. more than 7) should be eliminated. Compounds that do not have at least one oxygen or one nitrogen atom must be eliminated. Some filters also eliminate molecules that contain "unusual" atoms such as P, Se, Si, B, Metals, As etc...
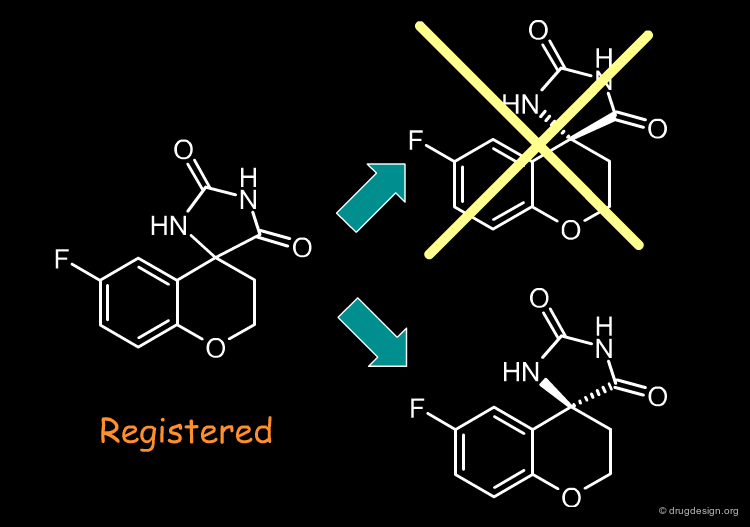
Remove Molecules with Multiple Chiral Centers¶
Molecules with more than one chiral center are usually eliminated, because of the potential for enantiomerically complex mixtures and increased analytical needs. The significance of addressing stereospecificity in the early stages of drug discovery is not widely appreciated, however this might change: advanced combinatorial chemistry and advantages in terms of patent position might prove the chirality issue to be beneficial.

Tailor-Made Filtering¶
Beyond to his pioneering contribution, Lipinski realized the limitations of his model: there are many exceptions, including for example substrates for transporters and natural products. Nowadays, the refinements are not so directed towards a global model; instead, rules are developed in a tailor-made manner, for specific zones such as CNS, anti-tuberculosis drugs etc...

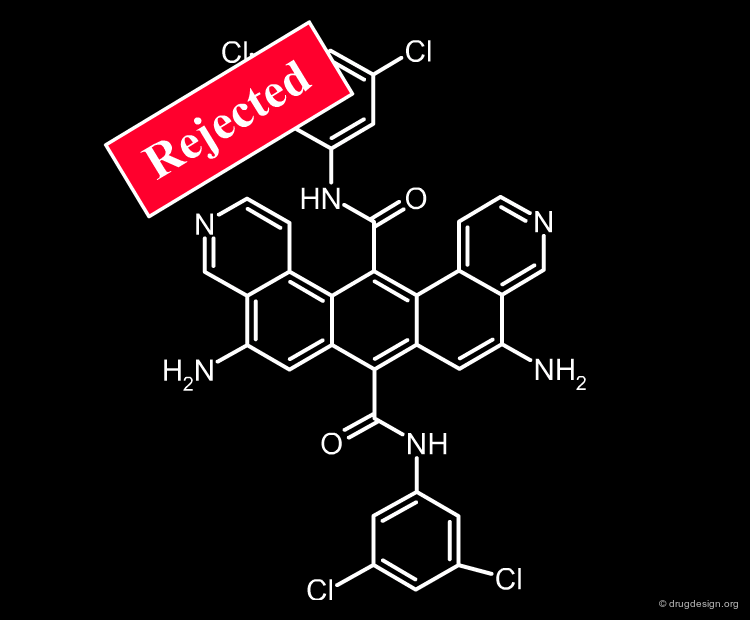
Assisting Medicinal Chemist Expertise and Intuition¶
Structural filtering carries a risk, which is entirely the responsibility of the medicinal chemist. He decides to broaden or to restrict any of the structural filters previously presented; he may also include or exclude compounds based on value judgments (e.g. business, intellectual property reasons). He is fully committed to the discovery of a good drug candidate and his efficiency must be increased by sophisticated tools that assist him in making the right decision. Although rejected by all state of the art filters used in drug discovery, a structure as for example the one shown below, can still be kept if the medicinal chemist decides to do so!

Privileged Drug-Like Scaffolds¶
Along the same "drug-like" perspective, analyses were also made in terms of the more frequent scaffolds encountered in known drugs. The 32 most commonly occurring frameworks in drugs are presented below (three scaffolds per page). Note that these are not molecules but templates, with nodes being either carbon atoms or heteroatoms.
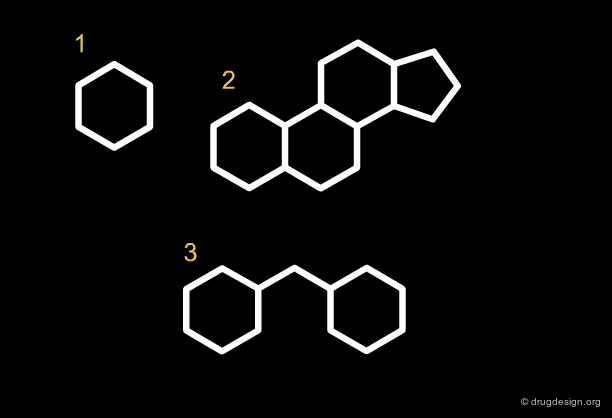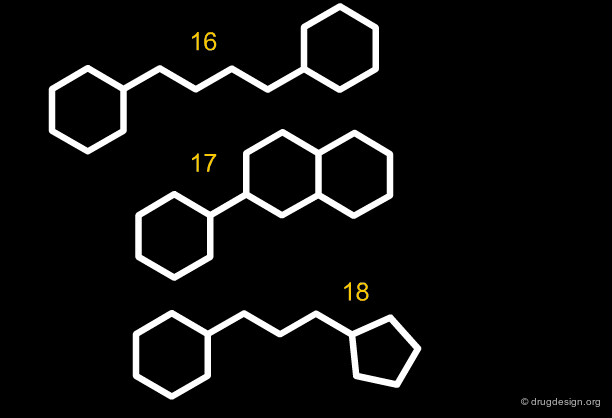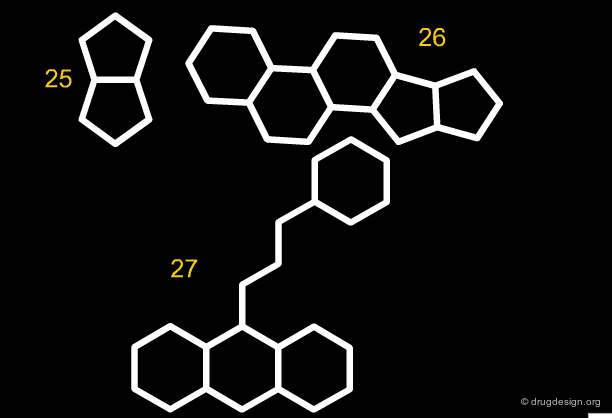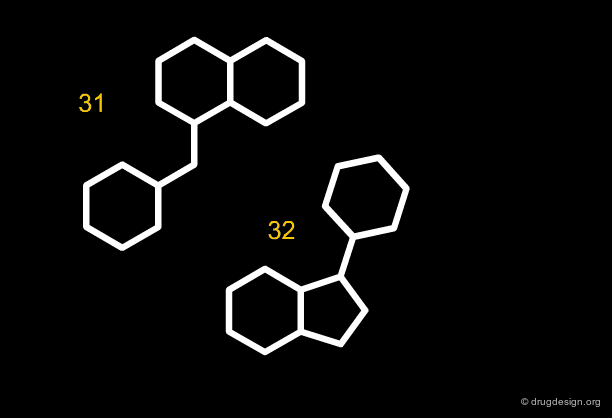
articles
Prediction of "drug-likeness" W.P. Walters et al. Advanced Drug Delivery Reviews 54 2002
Methods for Drug Discovery: Development of Potent, Selective, Orally Effective Cholecystokinin Antagonists Evans, B. E.; Rittle, K. E.; Bock, M. G.; DiPardo, R. M.; Freidinger, R. M.; Whitter, W. L.; Lundell, G. F.; Veber, D. F.; Anderson, P. S.; Chang, R. S. L.; Lotti, V. J.; Cerino, D. J.; Chen, T. B.; Kling, P. J.; Kuntel, K. A.; Springer, J. P.; Hirshfield. J. J. Med. Chem. 31 1988
Privileged structures-an update Patchett, A.; Nargund, R. P. Annu. Rep. Med. Chem. 35 2000
Privileged molecules for protein binding identified from NMR-based screening Hajduk, P. J.; Bures, M.; Praestgaard, J.; Fesik, S. W. J. Med. Chem. 43 2000
The combinatorial synthesis of bicyclic privileged structures or privileged substructures Horton, D. A.; Bourne, G. T.; Smythe, M. L. Chem. Rev. 103 2003
Medicinal chemistry of target family-directed masterkeys Muller G. Drug Discov Today 8 2003
Privileged Structures: Applications in Drug Discovery DeSimone, R. W.; Currie, K. S.; Mitchell, S. A; Darrow, J. W.; Pippin, D. A. Comb. Chem. High Throughput Screen. 7 2004
Privileged structures as leads in medicinal chemistry Costantino, L; Barlocco, D. Curr. Med. Chem. 13 2006
Building Blocks Based on Known Drugs¶
Following the drug-like approach, suppliers of building blocks propose libraries constructed on the scaffold of a known reference drug. These libraries are presented with the corresponding reference drug, the therapeutic area, the CAS code, the most important ADME-related parameters (calculated or experimental), to provide evidence that in the first place, the structures proposed are not bad ones.

The Computational Approach¶
The mechanisms involved in pharmacokinetics are governed by complex physiology and biochemical processes, and Lipinski-like rules are not refined enough. The computational approach to the prediction of pharmacokinetic and ADME characteristics is one of the newest techniques developed in pharmacokinetics and toxicity. The aim is the prediction of these properties for a potential drug molecule, although it only exists as a virtual structure.

Prediction of Absorption¶
Computational models for predicting intestinal absorption have started to be of practical utility (e.g. in CACO-2 and MDCK cells) as evidenced by the replacement of the cellular permeability screens by computational models in some companies. Commercial software such as IDEA (TM) and GastroPlus (TM) have been proven to give correct predictions of intestinal absorption in 70% of the cases. However, predicting oral absorption remains a difficult task and in-silico methods are still in their infancy.

book
Sinko et al. Reliability and robustness of selected commercial programs
1993
Prediction of Metabolism¶
Ideally, in-silico tools must predict most probable metabolic routes, binding to metabolizing enzymes, rate of metabolism, possible metabolic drug-drug interactions etc... Computer programs have been developed (e.g. METEOR, METABOLEXPERT, QDIP) that can help to design clinically relevant in vivo studies, but cannot replace in vivo investigation. Metabolism still remains very difficult to evaluate and to predict. Show below is the interaction of a drug (warfarin) with a Cytochrome P450 enzyme.

Prediction of Distribution and Elimination¶
Tissue distribution is one of the most important determinants of the pharmacokinetic properties of a drug. The computational approach aims at predicting the tissue distribution of a compound before animal or clinical trials are made. Indeed, early predictions of the volume of distribution (at steady state) and the tissue distribution of a compound have proven to be feasible, and are used in the pharmaceutical industry. Up to now little work has been done in the computational modeling or prediction of elimination.

Prediction of Toxicity¶
The prediction of toxicity is an extremely challenging problem because of the multiplicity of toxicity mechanisms. Particular attention is paid to the prediction of toxicological liabilities, such as carcinogenicity, mutagenicity, hepatotoxicity and teratogenicity. Computational toxicity methods are either knowledge-based expert systems (e.g. DEREK), or based on statistically-oriented methods (e.g. TOPKAT).
Lack of Standardized ADMET Databases¶
Computational modeling approaches to the prediction of ADMET properties aim at the early identification of compounds with unfavorable pharmacokinetic and ADME properties. Such methods should also allow for the rational optimization of a series, where compounds with improved properties are designed and assessed beforehand. Large databases of ADMET data associated with structures are needed to build computational models to link structural changes with changes in response. The lack of such databases has particularly hampered the development of this field, which is fairly complex.
Available Software¶
Examples of programs developed to predict ADMET properties or to help in designing animal or clinical experiments are shown below. Click the names of the software suppliers to see the different programs that are available.

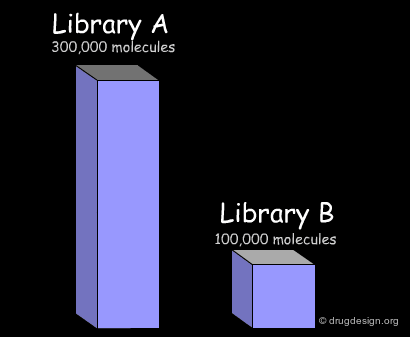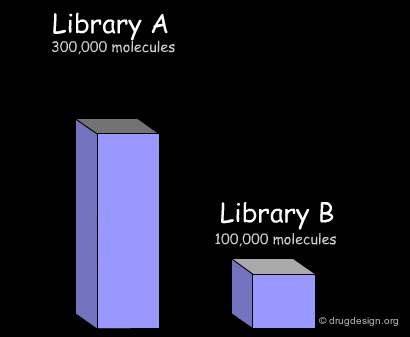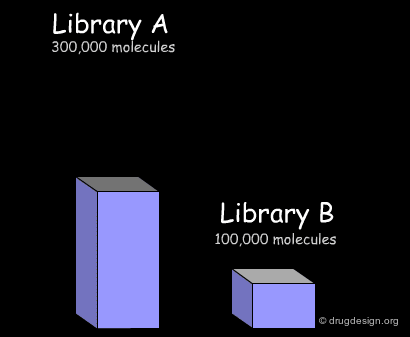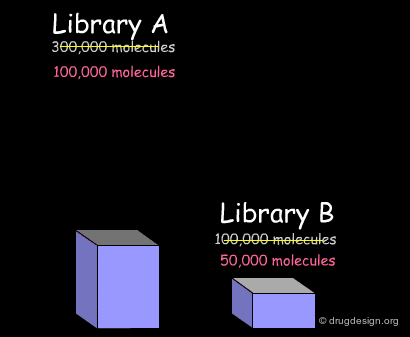
Example of Library Analysis¶
Common Treatments in Library Analysis¶
Here we present common treatments in library analysis. We show how the data are treated and how the results are assessed, by comparing two libraries. Beyond the example, the following aspects should be addressed: which library is the most interesting? which one is the most diverse? and other questions that are raised when building a library for screening applications.

Import of Libraries in a Common Database¶
The first step consists of importing the two libraries to be compared into a common database, so that the molecules can be processed by the analysis softwares. Generally the suppliers of libraries provide the databases (in the SDF format) through the web. Once a download is executed, the corresponding library is automatically imported.

Cleaning Up the Database¶
The first treatment consists of cleaning up the database. For example remove duplicate molecules, remove compounds that are ill defined or with errors (e.g. C+, C-, O+ or N- atoms with wrong valence) and also remove salts and counter ions. These are the most common treatments needed for preparing the data for subsequent analyses.

Set Stereoisomers¶
Often, the stereochemical information of chiral molecules is not specified (e.g. racemic mixtures). In which case and to simplify the analysis only one stereoisomer is kept with an arbitrary stereochemistry assigned at random, instead of enumerating the various possibilities. The consequences of this choice are not too serious, when considering only the 2D aspects (this would not be suitable for 3D treatments such as for example 3D searching).
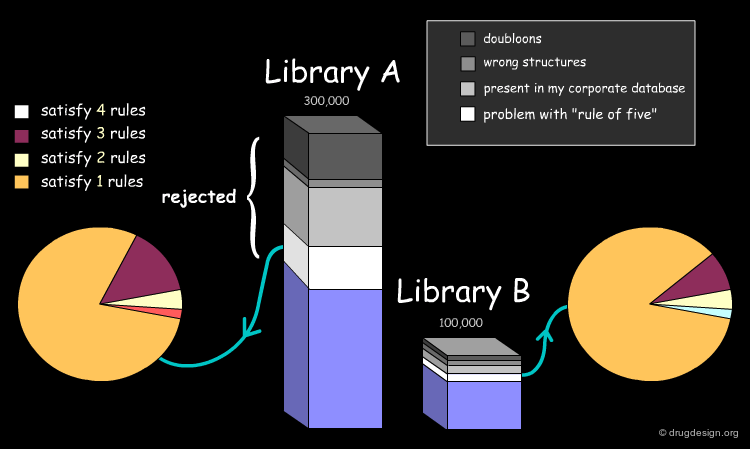
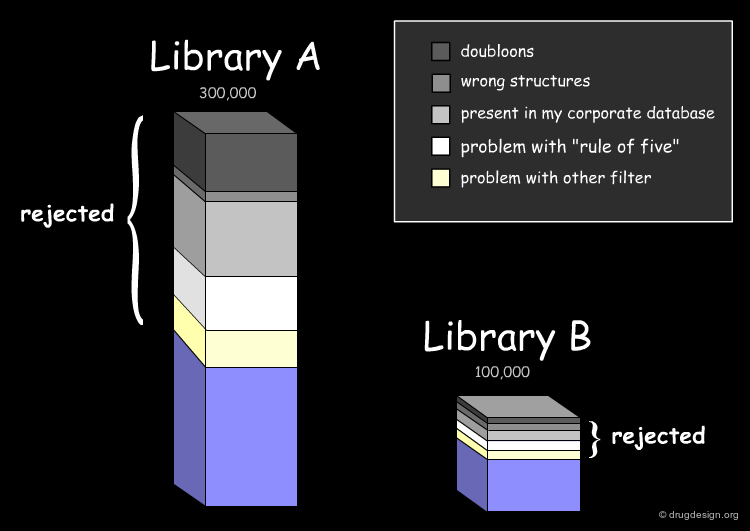
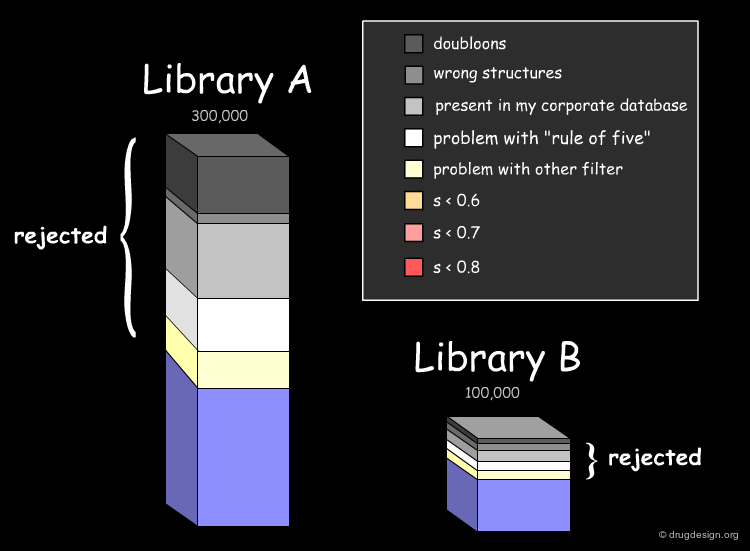
Assessing the Uniqueness of a Library¶
It is essential to assess one's interest in a given library (e.g. A or B) and have the means for comparing it to the collection of compounds currently used (e.g. corporate database). Detailed analyses can be made with overall conclusions visually represented. In particular the analyses indicate how many novel compounds are present in the library, the number and the reasons for which other compounds were rejected.

Drug-Likeness: Lipinski Rule of 5¶
The fraction of compounds which are consistent with the drug-likeness "Lipinski rules" criteria is evaluated (it provides a guide for determining if a compound can be orally bioavailable). It is useful to know how many compounds of the library examined satisfy all Lipinski rules or some of them.
Drug-Likeness: Other Filters¶
The next step in the evaluation of the drug-likeness of the library is to consider other properties recognized as important, such as the number of rotatable bonds, the number of rings and their size, and also the presence of undesirable functional groups (e.g. chemically reactive moieties, toxicophores) etc...
Diversity Analysis with Molecular Descriptors¶
The analysis of the molecular diversity of libraries A and B can be calculated using fast 2D molecular descriptors characterizing specific properties such as hydrophobic and hydrophilic effects, polarizability and electrostatic interactions. Using normalized descriptors and similarity indices between compounds of each database (calculated for example with the Tanimoto equation) diversity patterns can visualize the degree of similarity of the compounds of a given database (small values mean large dissimilarities).
Diversity Analysis with Fingerprints and PCA¶
Another method for diversity analysis consists of calculating a number of structural keys for each compound and reducing them with principal component analysis (PCA). The compounds can be then displayed on a graph using the first latent variables (in the example below, 150 structural keys were used and reduced to two variables). In this way, the diversity and the location of each database in the chemical space can be compared graphically.

Final Results¶
In the example presented here, we discussed the successive steps for the comparative analysis of two libraries A and B. We illustrated the use of structural keys for assessing their diversity (with molecular descriptors or structural keys). Then, we showed how to reduce the complexity (dimensionality) of the analyses by using PCA methodologies. Finally, from the initial set of compounds initially gathered, we were able to reject 50% of them, and identify unique compounds with suitable properties which deserve to be included in the corporate collection of compounds, and represent a valuable library for virtual screening applications.
Copyright © 2024 drugdesign.org