Cheminformatics: Principles and Applications¶
Info
Cheminformatics is an inter-disciplinary field involving chemistry, physics, mathematics, computer science and information technologies that applies informatics methods to solve chemical problems. This chapter presents the principles and applications of cheminformatics. Pharmacophore mapping is presented in the molecular modeling section; molecule searching and reaction searching are presented in the section on chemical information systems, and an introduction to QSAR modeling is given in the section on data analyses.
Number of Pages: 229 (±5 hours read)
Last Modified: May 2009
Prerequisites: None
Introduction¶
What is Cheminformatics ?¶
Cheminformatics is a rapidly growing field that appeared in the late 1990s. It is an inter-disciplinary field involving chemistry, physics, mathematics, computer science and information technologies. No consensus has been reached for defining the new discipline and a rather broad definition is used: "cheminformatics applies informatics methods to solve chemical problems".

articles
Prediction methods and databases within chemoinformatics: Emphasis on drugs and drug candidates Svava Osk Jonsdottira et al. Bioinformatics 15(21) 2005 10.1093/bioinformatics/bti314
The central role of chemoinformatics Johann Gasteiger Chemometrics and Intelligent Laboratory Systems 82 (1-2) 2006 10.1016/j.chemolab.2005.06.022
** ** K. Brown Annual Reports in Medicinal Chemistry 33 1998
Chemistry plans a structural overhaul E. Russo Nature 419 2002 10.1038/nj6903-04a
book
J. Gasteiger and T. Engel Chemoinformatics, A Textbook Weinheim: Wiley 2003
Tudor I. Opera (Ed.) Series Methods and Principles in Medicinal Chemistry Wiley-VCH Verlag GmbH 2005
wikipedia
Cheminformatics or Chemoinformatics ?¶
Although the term first used was "chemoinformatics" and a group of European academic researchers argued to keep it in 2006, the term "cheminformatics" is used more frequently. The graph below gives the number of hits from Google for the two terms per country.

book
J. Gasteiger and T. Engel Chemoinformatics, A Textbook Weinheim: Wiley 2003
J. Bajorath
Totowa: Humana Press
A. R. Leach and V. J. Gillet
Dordrecht: Kluwer Academic Publishers 2003
Cheminformatics and Drug Discovery¶
One particular focus of cheminformatics is drug discovery, where tools and new methods are developed "to assist discovery scientists in their decisions about what to test and make next" (Lahana 2002). Cheminformatics has completely changed the face of drug discovery and optimization in the last decade.
articles
Cheminformatics - decision making in drug discovery Roger Lahana Drug Discovery Today 7 (17) 2002 10.1016/S1359-6446(02)02368-1
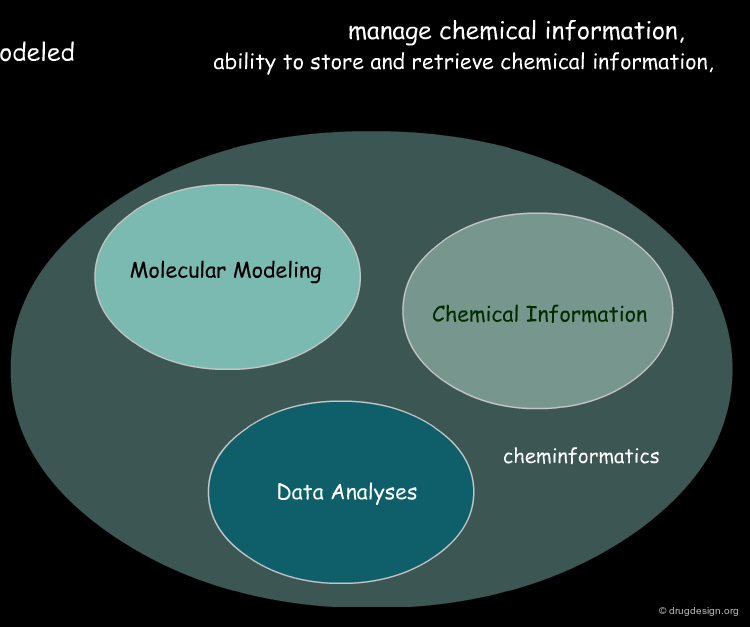
Cheminformatics: Integration of Three Disciplines¶
Although it emerged as a super-discipline in the late 1990s, cheminformatics is the result of integrating old well-established disciplines: molecular modeling, chemical information and data analysis. Molecular modeling is a field that models molecular behavior, chemical information deals with chemical information, and data analysis is a field that develops methodologies for the analysis of data to extract useful information.
Historical Background of Pharmaceutical Research¶
To better understand the origin of cheminformatics it is worth looking at the major milestones in the history of the pharmaceutical industry over the last decades. The overview below explores the context which led to the birth of the new discipline.
Media
This picture was made using the QuteMol Developed by Marco Tarini and Paolo Cignoni of the Visual Computing Lab at ISTI - CNR
Molecular Modeling¶
The 1980s were marked by developments in molecular modeling. Scientists started to apply models to understand molecular properties: mathematical models to understand structure-activity relationships, 3D models to understand molecular interactions, force-fields to understand molecular energies, and pharmacophore models to understand the structural features of molecular recognition. The first computer programs that were developed were reserved for specialists: the molecular modelers.

Chemical Information¶
At the same time another discipline known as "chemical information" emerged. This field involves the collection, classification, manipulation, storage, and retrieval of information associated with chemical compounds. This discipline was closer to the activities of traditional medicinal chemists, and often more useful in their day to day work.

wikipedia
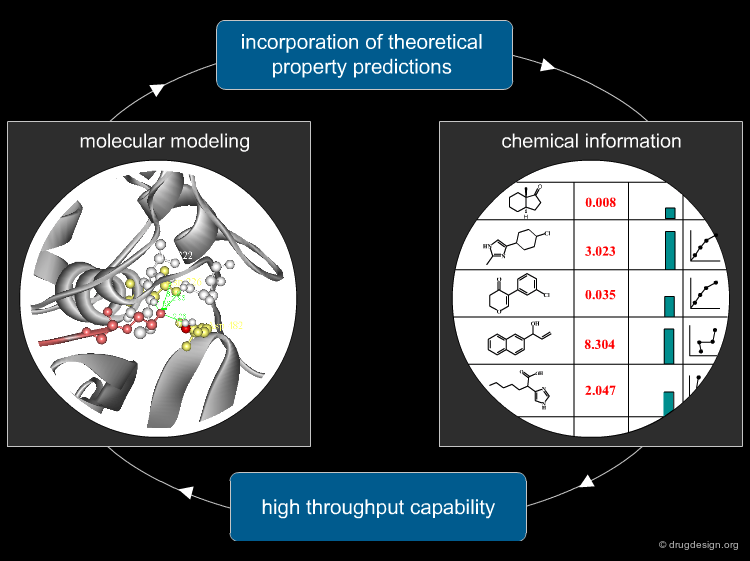
Coupling Modeling and Chemical Information¶
With the vast quantities of data produced by drug research in the early 1990s, molecular modeling had to move from manual modeling to automated methods. Modeling programs started to be coupled with chemical information. Chemical information opened the high-throughput dimension to modeling, and molecular modeling enabled chemical information to incorporate predicted properties into the databases.
The Data Analysis Contribution¶
With the high throughput chaos triggered in the early 2000s by high throughput screening and combichem, Data Analysis was added to modeling and chemical information to form cheminformatics. Data Analysis consists of the application of the laws of probability and statistics to transform raw data into useable information. It generates knowledge from the data, guides rationales and helps formulate new hypotheses. Data analyses provide effective decision-support tools that are critical in drug discovery.

Example of Successful Integration¶
The molecule shown below represents an example of discovery that exploited chemical information (database searching), molecular modeling (conformational analyses) and data analyses (similarity analyses). The molecule is a potent inhibitor of the vascular endothelial growth factor tyrosine kinase receptor KDR.

articles
Identification of a New Chemical Class of Potent Angiogenesis Inhibitors Based on Conformational Considerations and Database Searching Pascal Furet, Guido Bold, Francesco Hofmann, Paul Manley, Thomas Meyer and Karl-Heinz Altmann Bioorganic and Medicinal Chemistry Letters 13 2003 10.1016/S0960-894X(03)00626-7
Definitions of Cheminformatics¶
The difficulty in reaching a consensus for defining cheminformatics stems from the historical context described in the previous pages. This resulted in different definitions for cheminformatics depending on the field of expertise of the person defining it. With a rather broad definition, people in different disciplines can feel comfortable with it.

articles
Prediction methods and databases within chemoinformatics: Emphasis on drugs and drug candidates Svava Osk Jonsdottir et al. Bioinformatics 15(21) 2005 10.1093/bioinformatics/bti314
Chemoinformatics, what it is and how does it impact drug discovery Brown, F.K. Annual Reports in Medicinal Chemistry 33 1998
Cheminformatics vs. Structural Bioinformatics¶
Cheminformatics, which deals essentially with the study of small molecules, is the central component in ligand-based drug design. With the explosion of macromolecular structure determination in the 1990s, a new discipline emerged which is known as "structural bioinformatics" that deals with the 3D structure of macromolecules. Structure-based drug design is the outcome of the encounter between cheminformatics and structural-bioinformatics.

wikipedia
Encoding Molecules¶
In order to manipulate molecules with computers, we need to define a way to encode molecules. This includes the encoding of chemical structures in 1D, 2D and 3D, chemical reactions, molecular surfaces and volumes, molecular properties and descriptors. This will be presented in the chapter entitled "Encoding Molecules".

Development of Algorithms¶
Cheminformatics is part of computer science where programming and algorithms are of key importance. In order to solve chemical and drug discovery problems, cheminformatics scientists develop new approaches, implement new codes, create and maintain chemical databases. Modern programming and algorithms constitute an important part of the syllabus of cheminformatics courses. Examples of essential algorithms used in cheminformatics are presented later in this chapter.

Facilitate Multidisciplinary Communication¶
One of the keys that enabled the successful integration of these different disciplines was the development of software facilitating multidisciplinary communication. For example, the software development pipeline shown here enables the chemist to create data flows, execute tasks in modeling or in chemical informatics, and then to analyze the results interactively. It places tools in the hands of chemists that were once solely reserved for experts.
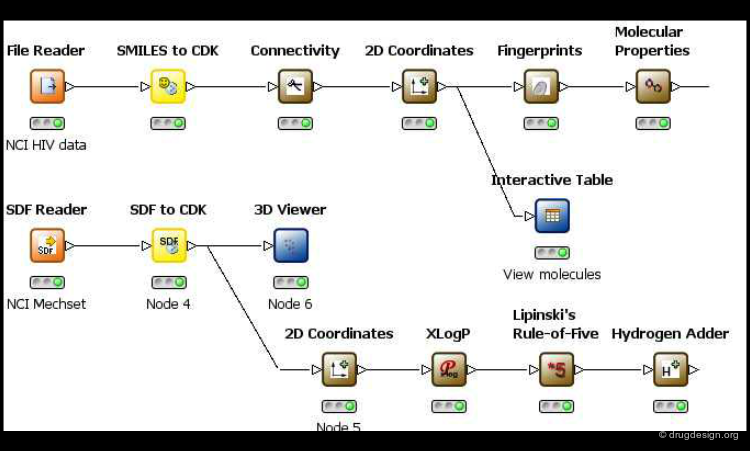
Media
screenshot from KNIME Launch PDB Ligand Explorer
Molecular Modeling¶
Pharmacophore Mapping¶
Pharmacophore mapping is a powerful and essential component in drug discovery. It attempts to understand and exploit the structural features of molecular recognition. In the absence of the 3D structure of the target protein, it is the only way to rationally design new drugs. It has been used exclusively for more than 30 years, in a number of projects. With the explosion of X-ray protein determinations, this approach continues to be largely used.

wikipedia
The Concept of 3D Pharmacophores¶
A pharmacophore is a specific 3D arrangement of chemical groups in a molecule which is essential to its biological activity. The official IUPAC definition is: "a pharmacophore is the ensemble of steric and electronic features that is necessary to ensure the optimal supramolecular interactions with a specific biological target structure and to trigger (or to block) its biological response".

articles
Glossary of terms used in medicinal chemistry (IUPAC recommendations 1998) C. G. Wermuth, C. R. Ganellin, P. Lindberg And L. A. Mitscher Pure and Appl. Chem 70 (5) 1998 10.1351/pac199870051129
Pharmacophoric Structural Elements¶
Efforts are being made to describe the structural elements of a pharmacophore in abstract bioisosteric terms such as hydrophobic, H-bond donors and acceptors, positively and negatively charged groups etc.. The relationships between the different elements of a pharmacophore are described in terms of distances and angles.

What is Pharmacophore Mapping ?¶
Pharmacophore mapping is a computerized approach used to derive 3D pharmacophores based on the 3D alignment of the geometric and physicochemical features of known active reference compounds.

book
Thierry Langer (Editor), Remy D. Hoffmann (Editor) Series: Methods and Principles in Medicinal Chemistry Wiley 2006
Osman F. Guner
International University Line, La Jolla 2000
Manual Pharmacophore Mapping¶
Before the advent of computers, medicinal chemists used to search for pharmacophoric patterns by measuring distances with a ruler on Dreiding models. This process requires good chemical intuition (ability to guess which groups to consider) and theoretical knowledge (using acceptable conformations).

book
C. G. Wermuth
Academic Press 1996
Derivation of Pharmacophore Hypotheses¶
The derivation of pharmacophore hypotheses is done by applying a computer algorithm to a training set of molecules with known activities. The goal is to identify rules that govern the system: What are the common features shared by the active molecules? What structural elements are essential for the activities? Which are detrimental? The steps involved in the derivation of the pharmacophore are presented in the following pages.

Steps in Deriving a Pharmacophore¶
The construction of a pharmacophore requires several steps: (1) the selection of the training set; (2) the generation of the conformers for all molecules; (3) the assignment of pharmacophoric elements (annotation) of all the conformers and (4) finding the best overlap of pharmacophoric elements. The principle is very simple but in practice it poses problems that still continue to mobilize the efforts of today's generation of computational chemists.

book
Thierry Langer (Editor), Remy D. Hoffmann (Editor) Series: Methods and Principles in Medicinal Chemistry Wiley 2006
Osman F. Guner
International University Line, La Jolla 2000
The Initial Training Set¶
The first step in pharmacophore mapping is the selection of a training set, from which it is possible to derive hypotheses. The molecules must act according to the same mechanism of action. The ideal dataset consists of many active molecules that are not too flexible. When chemically related molecules are in the initial dataset, pairs of active and inactive analogs may be of high informational content.

book
Thierry Langer (Editor), Remy D. Hoffmann (Editor) Series: Methods and Principles in Medicinal Chemistry Wiley 2006
Osman F. Guner
International University Line, La Jolla 2000
Generation of Conformers¶
Since molecules are flexible, each molecule in the dataset should be explored in its entire conformational space for a common pharmacophore arrangement in 3D. To avoid a conformational explosion, the presence of one rigid molecule in the training set may help restrict the conformational search for the others. If no rigid molecule is present, a systematic search can be first applied to one molecule, and the conformational space of the others will be restricted to the space of those already explored.

Which Combination of Structural Elements?¶
When the potential pharmacophoric elements have been annotated for all the conformers generated, the next step consists of finding the best combinations which correspond to a maximum overlap of pharmacophoric elements. This step can be done either manually or with automated methods.

Manual Method¶
In manual methods, a conformational analysis of the molecules is made, then a visual approach is used to determine which conformers exhibit the best overlap between all the molecules. Their 3D alignment reveals their common pharmacophore. A typical example is illustrated below; click on the different buttons to display the dataset, the conformers, the alignment and the common pharmacophore.
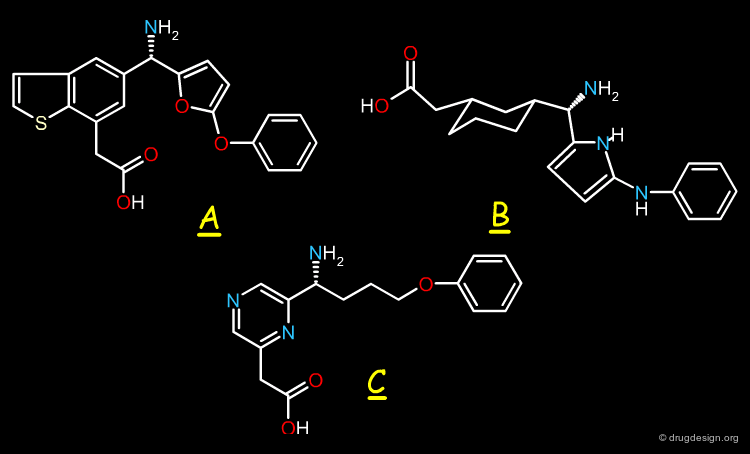



Example of Tricyclic Antidepressants¶
One of the first studies in pharmacophore mapping is Cohen's example of tricyclic antidepressants (1971). This study led to a simple pharmacophore model. The initial dataset, and the derived pharmacophore based on low energy conformations (ΔE < 1 kcal/mol), are shown below. The pharmacophore was further exploited to discover non-tricyclic structures.

articles
Drug Design in Three Dimensions Cohen NC Adv. Drug Res. 14 1985
Towards the Rational Design of New Leads in Drug Research N.C. Cohen Trends in Pharmacological Sciences 4 1983
Design of Non-Tricyclic Structures¶
Below (left view) is shown the rotatable superimposition of the molecules that led to the identification of the simple pharmacophore, visualized as balls. The right view shows one example of non-tricyclic molecule designed by the chemists, which proved to be a potent inhibitor of the reuptake of serotonin. This molecule appears in red in the 3D superposition.

articles
Drug Design in Three Dimensions Cohen NC Adv. Drug Res. 14 1985
Towards the Rational Design of New Leads in Drug Research N.C. Cohen Trends in Pharmacological Sciences 4 1983
Automated Methods¶
In automated methods the identification of common substructure matches is generally done with the clique algorithm. Other methods use the ensemble approach of distance geometry, genetic algorithms or maximum likelihood principles.

articles
Molecule-pharmacophore superpositioning and pattern matching in computational drug design G. Wolber, T. Seidel, F.Bendix and T. Langer Drug Discovery Today 13 (1-2) 2008 10.1016/j.drudis.2007.09.007
Automated Methods: the Conformational Issue¶
Catalyst and DISCO were the first programs developed for automated pharmacophore mapping. Conformational flexibility is addressed either by the pre-generation of a set of low energy conformers, or it is done on the fly, simultaneously with the matching algorithms.

Common Use of a Pharmacophore¶
Pharmacophore models can be used for many purposes; for example, they can be used to find hits, test hypotheses, understand structure-activity relationships, search for similarities with other molecules, design new lead molecules, create molecule libraries or generate ideas.
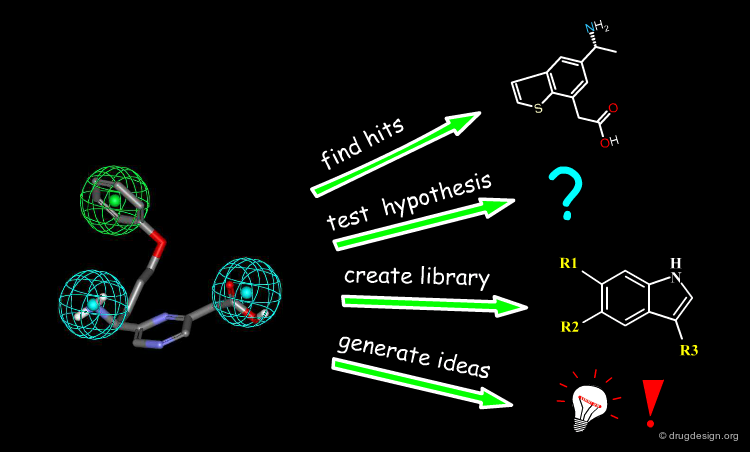
Pharmacophore Fingerprints¶
The most common use of a pharmacophore is to search 3D databases for molecules that contain the pharmacophore model. This is usually done by using 3D pharmacophore fingerprints. A pharmacophore fingerprint encodes specific pharmacophore arrangements in a binary key: 0 or 1 for a bit means that the 3D pharmacophore is absent or present in the structure.

Pharmacophore Databases¶
In the early 1990s, Golender et al. were the first to introduce the concept of pharmacophore databases. This was implemented in the APEX-3D software (commercialized by Accelrys) which enabled rapid ligand profiling. More recently, Inte:ligand has assembled a database of 3D pharmacophores consisting of 2500 pharmacophore models covering 300 clinically relevant targets.

book
Golender VE and ER Vorpagel In 3D QSAR in Drug Design: Theory Methods and Applications ESCOM Science Publishers 1993
Golender V, B Vesterman, O Eliyahu, A Kardash, M Kletzkin, M Shokhen and E Vorpagel In QSAR and Molecular Modelling: Concepts, Computational Tools and Biological Applications Prous Science Publishers 1995
Vorpagel ER and VE Golender Pharmacophore Perception, Development, and Use in Drug Design International University Line, La Jolla, CA 2000
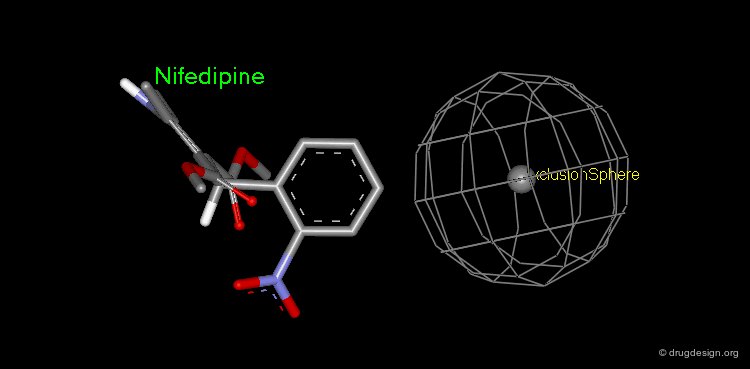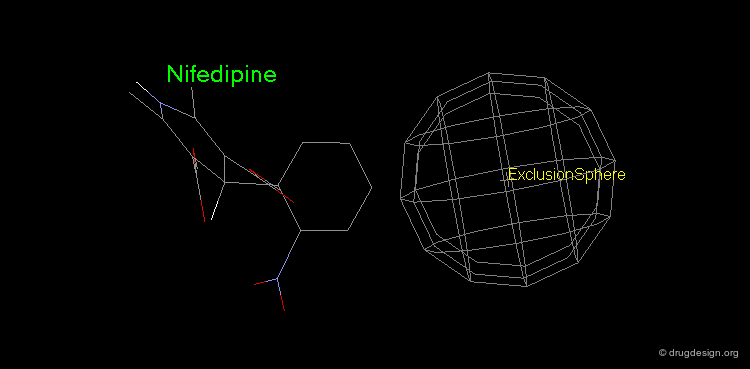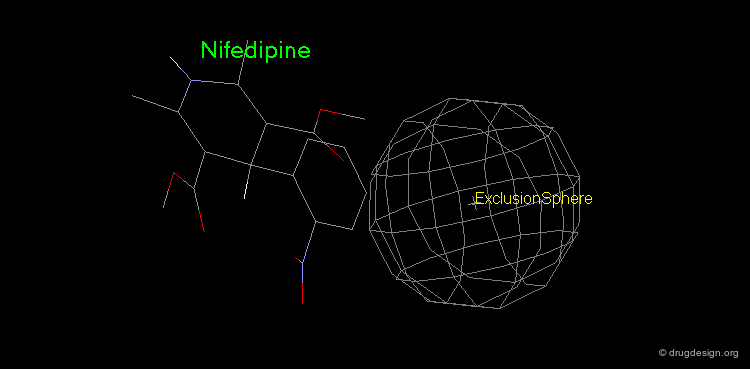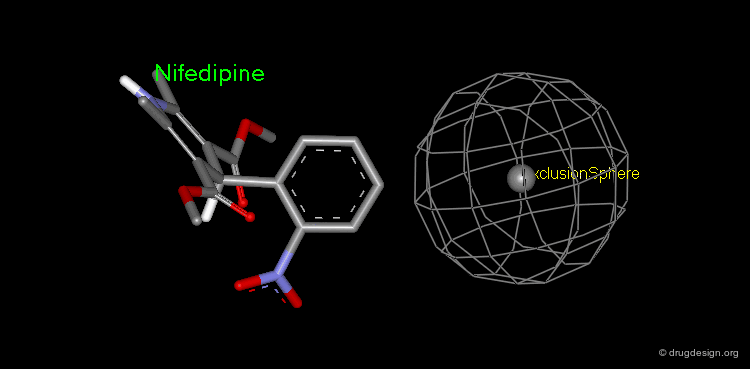
Combination with Other Methods¶
Pharmacophore mapping can be used in combination with other methods that contribute additional information. For example, exclusion spheres can be added to a pharmacophore when SAR studies reveal regions that should not be occupied by atoms of the ligands (probable bumps with the receptor). Shown below, the use of an exclusion sphere, based on the observation that para-substituted analogs in this series are always inactive. The database search will therefore identify hits that have no atoms or bonds in this region.
Combining Pharmacophore and Shape¶
Pharmacophore hypotheses can also be combined with shape constraints. The shape can be defined by that of a potent ligand or by the overlap of several active molecules. This makes it possible to exclude hits that stick out of this shape.

Structure-Based Pharmacophore Mapping¶
Recently, a radical change has taken place in pharmacophore mapping, with the development of structural bioinformatics. The new trend is called "structure-based pharmacophore mapping" and involves deriving pharmacophore models directly from the X-ray coordinates of complexes. This simplifies conformational and the pharmacophoric issues enormously because, the focus is now on relevant interactions derived directly from ligand-receptor complexes.
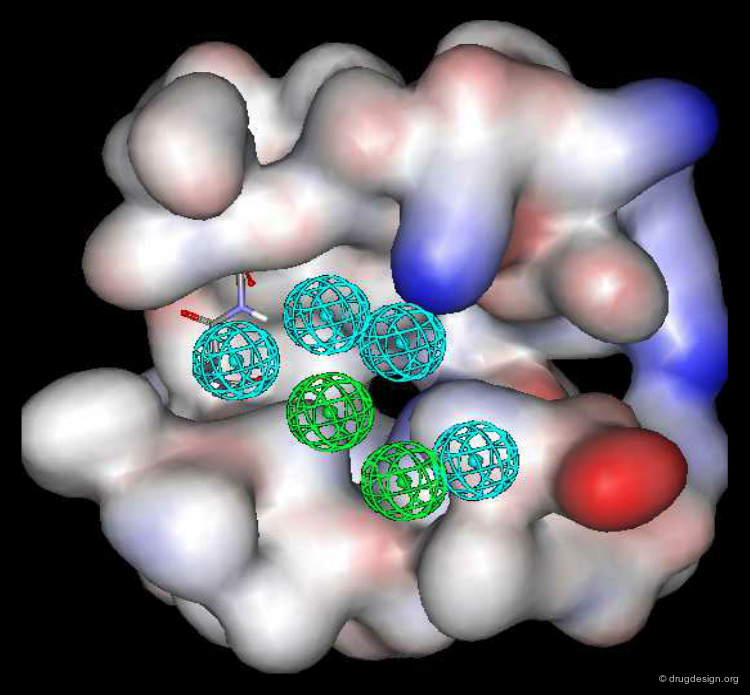
articles
Receptor-Based Pharmacophores for Serotonin 5-HT7R AntagonistssImplications to Selectivity Marcin Kolaczkowski, Mateusz Nowak, Maciej Pawlowski, and Andrzej J. Bojarski J. Med. Chem 49 2006 10.1021/jm060300c
Pharmacophore Based Receptor Modeling: The Case of Adenosine A3 Receptor Antagonists. An Approach to the Optimization of Protein Models Andrea Tafi, Cesare Bernardini, Maurizio Botta, Federico Corelli, Matteo Andreini, Adriano Martinelli, Gabriella Ortore, Pier Giovanni Baraldi, Francesca Fruttarolo, Pier Andrea Borea, and Tiziano Tuccinardi J. Med. Chem 49 2006
Three-dimensional models of histamine H3 receptor antagonist complexes and their pharmacophore Frank U. Axe, Scott D. Bembenek and Sandor Szalma J. Mol. Graphics Model 24 26 10.1016/j.jmgm.2005.10.005
A Pharmacophore Map of Small Molecule Protein Kinase Inhibitors Malcolm J. McGregor J. Chem. Inf. Model 47 2007 10.1021/ci700244t
LigandScout: 3-D Pharmacophores Derived from Protein-Bound Ligands and Their Use as Virtual Screening Filters Gerhard Wolber and Thierry Langer J. Chem. Inf. Model 45 2005 10.1021/ci049885e
Structure-Based Pharmacophore vs. Docking¶
Pharmacophore models describe the complex interactions between a target and its ligands in simple ways, making virtual screening based on pharmacophore queries very fast. One possible method is to start using structure-based pharmacophore mapping as a speedy low resolution method followed by more refined computationally high resolution docking.

The Ludi Program¶
One of the first programs in structure-based pharmacophore mapping was LUDI which searches for interaction centers in the protein and assembles potential new ligands by combining fragments from a three-dimensional structure library.

articles
The computer program LUDI: a new method for the de novo design of enzyme inhibitors Boehm, H. J. Journal of Computer-Aided Molecular Design 6(1) 1992 10.1007/BF00124387
LigandScout¶
LigandScout developed by Wolber et al. is a recent software tool that derives pharmacophores from structure-based complexes, and enables sophisticated pharmacophore analysis to create selective pharmacophoric screening filters for a specific target.
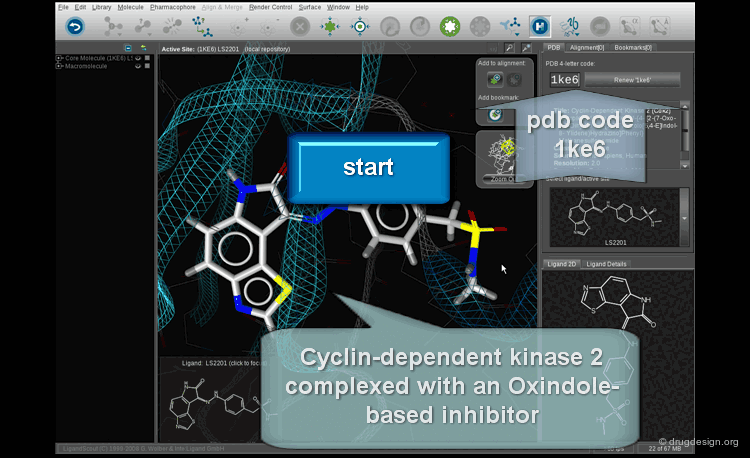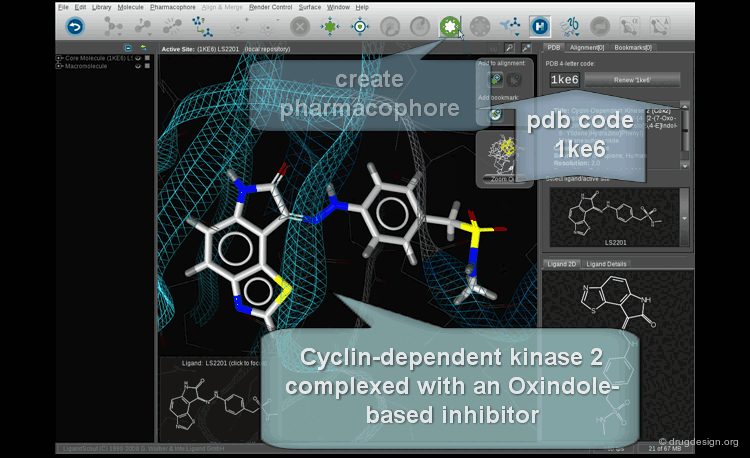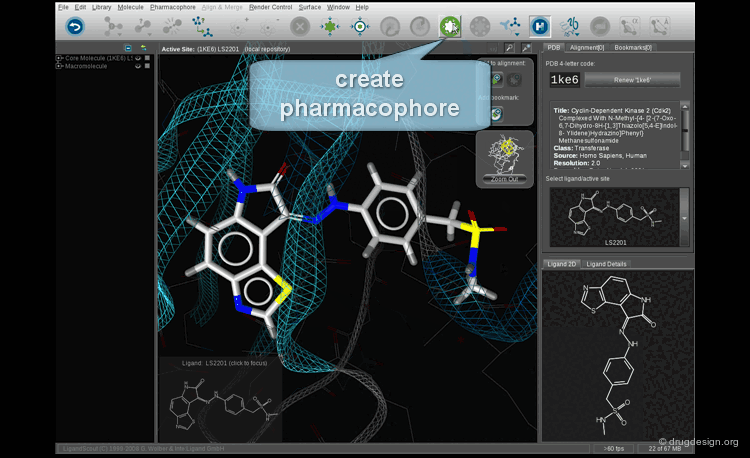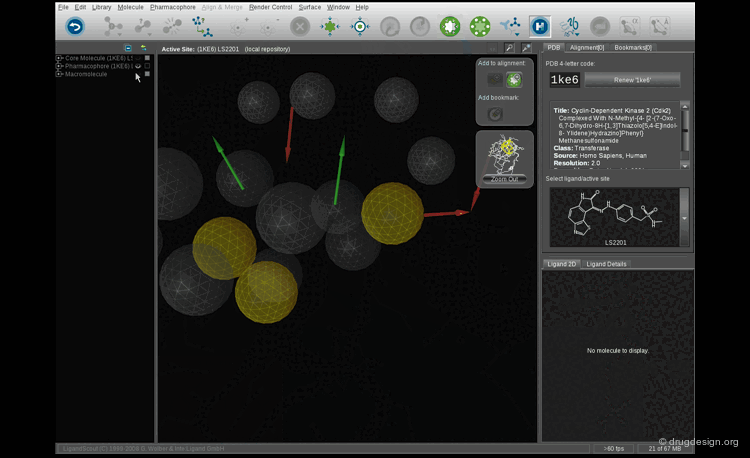
articles
LigandScout: 3-D Pharmacophores Derived from Protein-Bound Ligands and Their Use as Virtual Screening Filters Gerhard Wolber and Thierry Langer J. Chem. Inf. Model 45 2005 10.1021/ci049885e
Example of Pharmacophore Mapping¶
In order to discover novel 11β-hydroxysteroid dehydrogenase type-1 (11β-HSD1) inhibitors, Schuster at al. used a pharmacophore mapping methodology. In a subsequent step they searched for new inhibitors using the generated pharmacophores with a virtual screening approach. The work is summarized in the pages that follow.

articles
The Discovery of New 11-beta-Hydroxysteroid Dehydrogenase Type 1 Inhibitors by Common Feature Pharmacophore Modeling and Virtual Screening Daniela Schuster, Evelyne M. Maurer, Christian Laggner, Lyubomir G. Nashev, Thomas Wilckens, Thierry Langer, and Alex Odermatt J. Med. Chem 49 2006 10.1021/jm0600794
Initial Data Sets¶
Two training sets were used for the development of pharmacophore models: the first one consisting of selective 11β-HSD1 inhibitors, and the second one of non-selective inhibitors having both 11β-HSD1 and 11β-HSD2 inhibitory properties.

Pharmacophore Models¶
The Catalyst program gave several pharmacophore hypotheses for each training set. Two models (one for each training set) were retained; the corresponding representations (hypotheses 1 and 2) are shown below.

Exploitation of the Pharmacophores Generated¶
The pharmacophore models were exploited by virtual screening. Several hits were obtained, showing sub-micromolar activities for the inhibition of the 11β-HSD1 or 11β-HSD2 enzymes. Subsequent biological analyses revealed that compound 27 had interesting in vivo properties.

Programs for Pharmacophore Mapping¶
Most pharmacophore mapping programs are commercial; some of them are listed below.

Chemical Information¶
Molecule Searching¶
Introduced in the 1980s, Information systems are designed to store, search and retrieve molecules with their associated properties. They have transformed most facets of the field of chemistry, and especially the way chemists think and solve problems. Today they are routinely used across the entire spectrum of chemical activities, from analytical to synthetic chemistry.
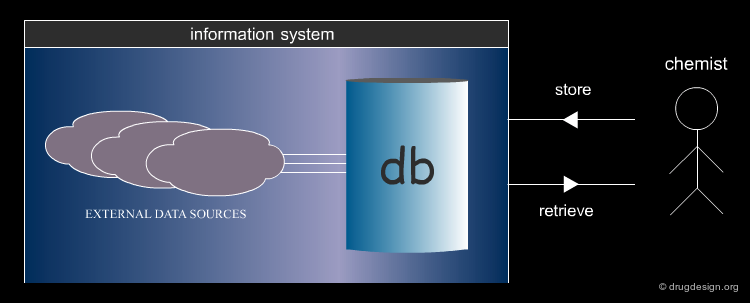
Components of an Information System¶
The major components of an information system are the following: a database to search, a query which defines what is looked for, a language to define the query, a computational mechanism to apply the query to the database of molecules, and a way to display, filter, save, or export the results ("hits").

Database Query Languages¶
Database query languages allow a user to interact with a database system. Structure Query Language (SQL) is the most widely used query language in modern relational database management systems (DBMS). It allows information specialists to query, update, delete information and control access to a database. Alternatively, most chemical information systems support graphical interfaces where the chemist can draw the query and introduce text, making these more adapted for non-specialists and end-users.

Media
Snapshot from eMolecules eMolecules
Quest for Information and Ideas¶
Although information systems can be used for a variety of purposes, we can classify them into two specific classes: (1) the search for information and (2) the search for new ideas. These two purposes will be developed further in the following pages.

Quest for Information¶
The first use of an information system is the extraction of information related to a specific compound. For example: who are the vendors, is the compound toxic, is it soluble and in which solvents, how it can be synthesized, what are its pharmacological properties etc... Technically the quest for information takes place in two steps: (1) the identification of the compound by using a query, and (2) the extraction of the desired information.

Identifying Compounds¶
Most databases of molecules are structured as a collection of records, organized in a relational model that can be viewed as a collection of tables, where information about molecules are represented in columns and rows. A key (also called a primary key) is used to uniquely identify a row (a molecule) in a table and establish relationships between tables. The aim of the query is to univocally identify this primary key. Some examples are illustrated in the following pages.


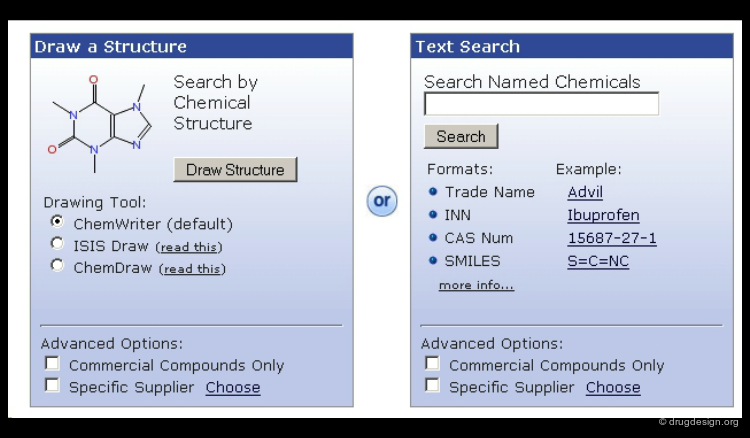
Searching by Name¶
Searching by name is the most trivial way to find a molecule in a database. Trade names, synonyms, CAS names, IUPAC names or systematic chemical names are typical input that can be used for the search.
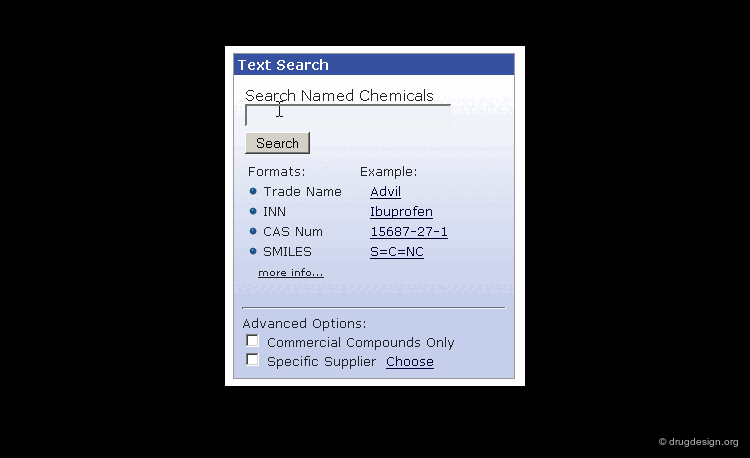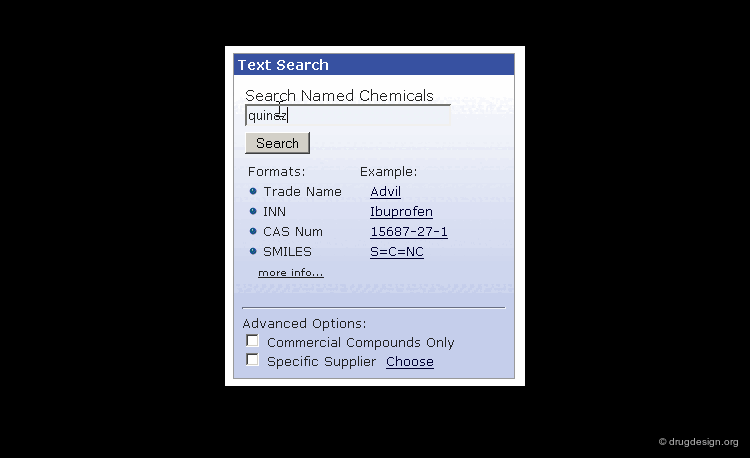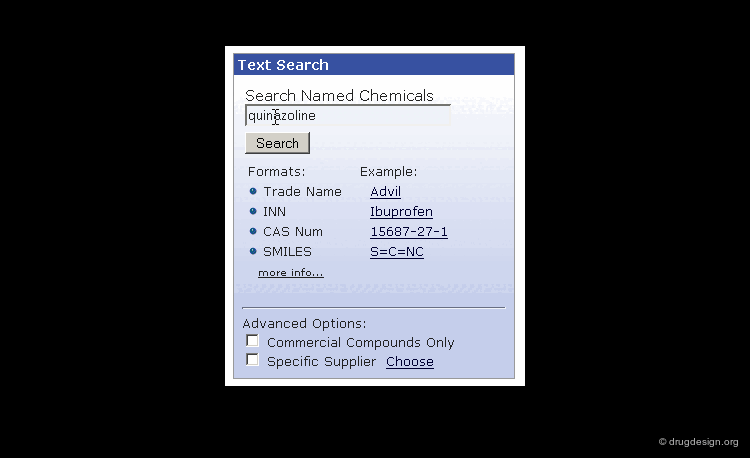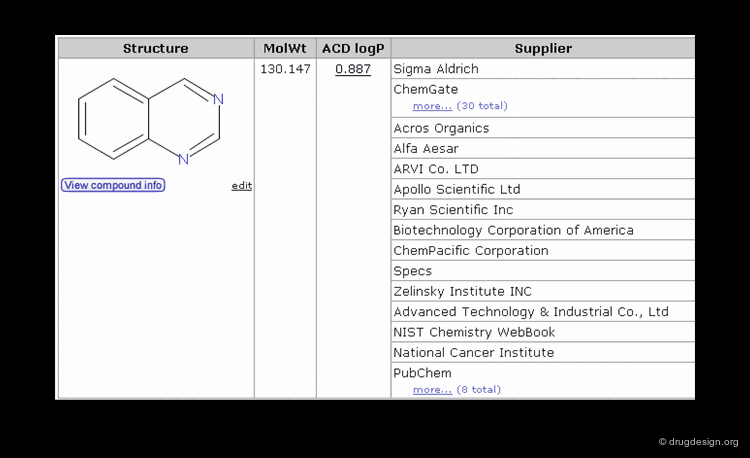
Media
snapshot from eMolecules eMolecules
# Problems when Searching by Name¶
A name search is often useful; however due to the multiplicity of possible names, a name search might not come up with the expected answer. Also, chemical names are often long and prone to typographical errors, introducing a high failure rate.

Searching by CAS Registry Number¶
The Chemical Abstracts Service REGISTRY is the largest and most current database of chemical substance information in the world. It has assigned CAS numbers (referred to as CAS-RN) to over 34 million compounds identified from the scientific literature from 1957 to the present, with additional substances going back to the early 1900s. For example the CAS-RN of taxol is 33069-62-4.
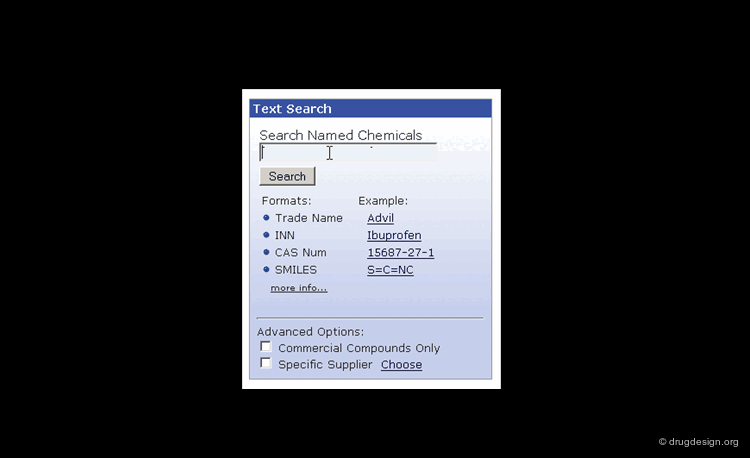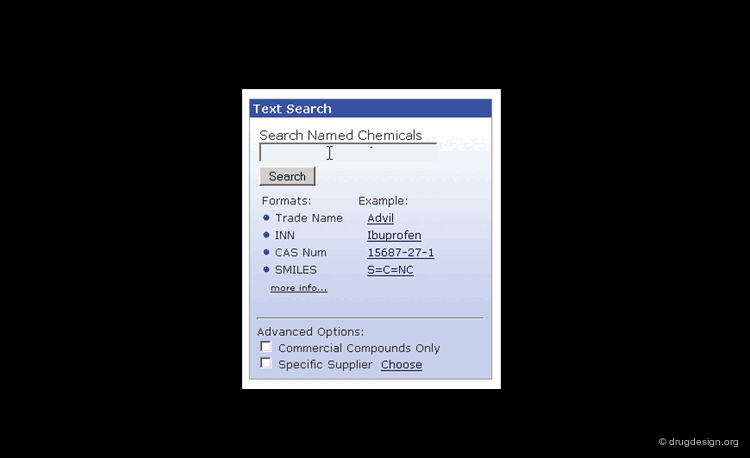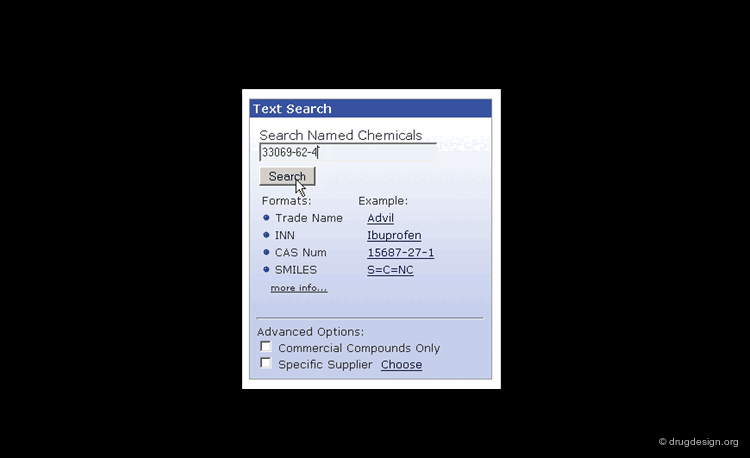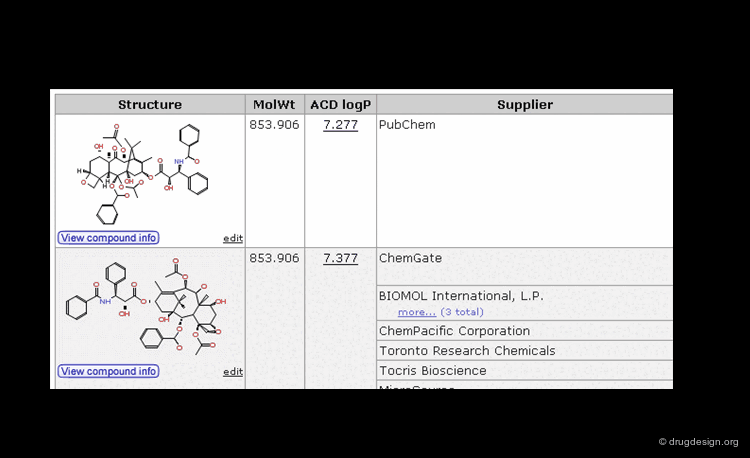
Media
snapshot from eMolecules eMolecules
Searching by 2D Molecular Structure¶
Searching by 2D structure is the most natural and efficient way to search for a molecule in a library. A 2D representation is unique and independent of any system of names or notation. It is much easier to use a graphical representation for drawing structures than to type complicated names or to use CAS numbers.

Media
snapshot from eMolecules eMolecules
Searching by SMILE String¶
SMILES (Simplified Molecular Input Line Entry System) is a line notation for representing molecules. It unambiguously describes the structure of a molecule and contains the same information as the connection table. Retrieving molecules using SMILES is a convenient way and a good alternative to searching by 2D molecular structure.

Media
snapshot from eMolecules eMolecules
Searching by Formula¶
It is always possible to search in a database by formulas; however, due to the great number of possible isomers, there might be many unwanted molecules in the hit list. The following molecules all correspond to the molecular formula C10H16O2.

Information Delivered by the Search¶
Once the molecule is identified, the related information is ready to be retrieved and displayed to the user. In addition to the inherent information stored in the database, some information systems have incorporated software that predicts molecular properties. This is the case of ChemSpider, a database that contains over 18 million chemical structures. The properties can be either calculated on the fly, or pre-calculated and saved in the database.

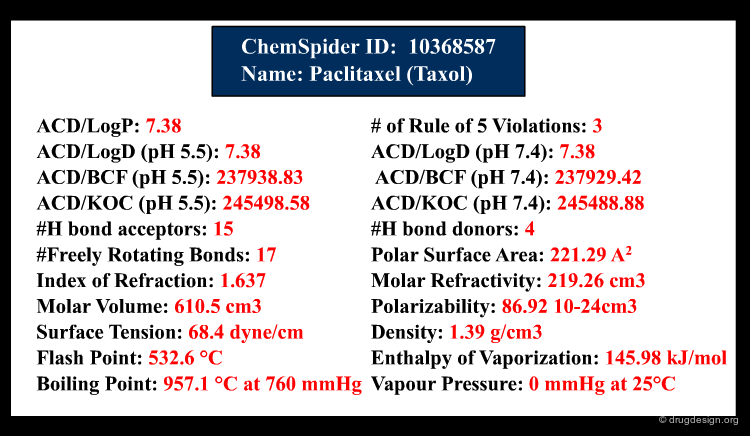
Types of Information¶
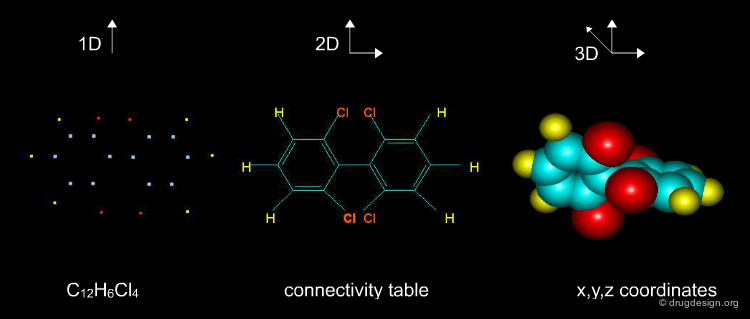
The information contained in a database is often classified in terms of 1D, 2D and 3D. 1D information is represented by a single value such as a molecular property (e.g. molecular weight, LogP, melting point etc.) or a text associated to the molecule (e.g. reference, therapeutic use, etc.). 2D information includes the information on the molecule connectivity, or other 2D information such as spectroscopic data (UV, IR, NMR). 3D information captures the three-dimensional coordinates or the spatial 3D properties of the molecule.

Media
Snapshot from ChemGate ChemGate
Quest for Ideas¶
The second use of an information system is a "search for new ideas". Contrary to the classical "search for information", which generates a definite result, the search for ideas involves an iterative process where the synergistic relationship between the chemist and the system accelerates the convergence towards a successful design. A good idea can rapidly generate promising hits and hits obtained in a search can lead to novel ideas. The search for new ideas can be done using either constrained or similarity searching.


Constrained Search¶
By gaining insights into the structure-activity relationships of the molecules, the chemist formulates working hypotheses. The incorporation of the knowledge acquired into well- formulated queries will help him find molecules to validate his hypotheses. The aim of this investigation is to restrict the search to a space of high informational content that is relevant to the hypotheses. This type of database searching is called a "constrained search".

Language to Define Constraints Associated to a Query¶
The difficulty in constrained searches is to translate the knowledge acquired about the molecules into intelligent queries for subsequent searches. Questions such as "is this hydrogen-bond essential?"; "do I need this tautomeric form?"; "can this atom be an oxygen?" etc..., are translated with a query language that specifies the constraints to be applied. Some components of constrained searches are presented in the following pages.

Define Constraints for Substituents¶
The good control of the substitution pattern is a key feature in the translation of knowledge gained with the molecules. For example for the series represented by the structure shown below, the SAR analyses indicate that ortho and meta-substituted analogs of the free phenyl ring have reduced activities, whereas the para substitution appears to be favorable. In this case the search should prevent meta and ortho substitution and accept any substitution in para.
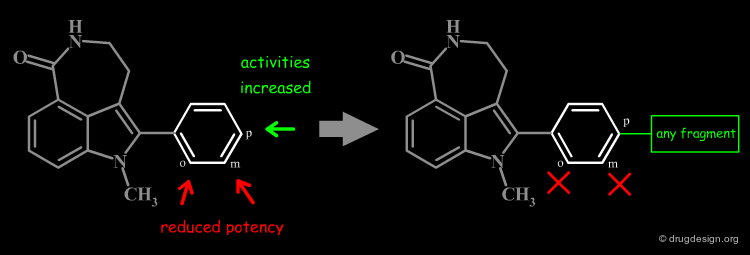
# Substituent Control by Explicit Hydrogens¶
By default, information systems accept all substitution patterns on a given atom, unless it is explicitly restricted. In the example of the previous page, to prevent substitution at the ortho and meta positions explicit hydrogen atoms are drawn, whereas the para position is left unrestricted, to accept any substitution at this position. This type of search is called "substructure search".

# Substituent Control by Substitution Numbers¶
An alternative of explicit hydrogens is the use of substitution numbers that define the total number of neighbors connected to the atom considered. For example in the ISIS system substitution counts such as s1, s2, s3 or s4 indicate that an assigned atom must have 1, 2, 3 or 4 neighbors.

Define Constraints for Atom Types¶
Constraints can be assigned for atom types and can be either explicitly defined (e.g. Bromine), or with a broader definition; in the ISIS convention "A" means any atom except hydrogen, "Q" means any atom except hydrogen or carbon. Moreover, a list of atoms can be given as desired, or not desired.

Define Constraints for Bonds¶
The definition of the constraints for a query bond can be either explicit (for example aromatic), or with a broader definition: for example "single/double", "double/aromatic". Double bonds can be defined "cis", "trans" or "cis/trans".

Define Constraints for Rings¶
Rings can be defined with constraints indicating their size.

Define Constraints for Stereochemistry¶
In the last ten years there has been increasing emphasis on the design of chiral molecules that bind with a high degree of stereoselectivity to biological receptors. Information systems have been designed to represent, define, store, search and retrieve molecules with specific stereochemistries.
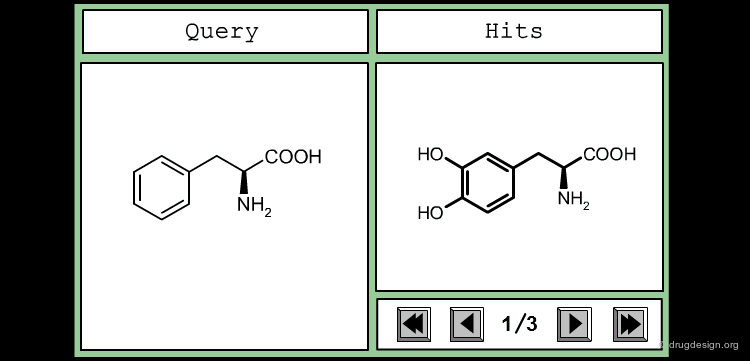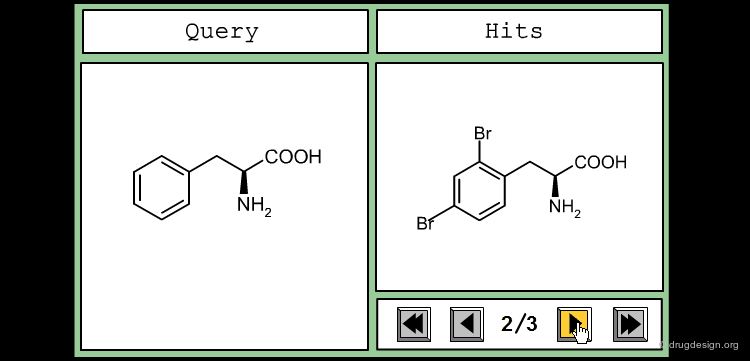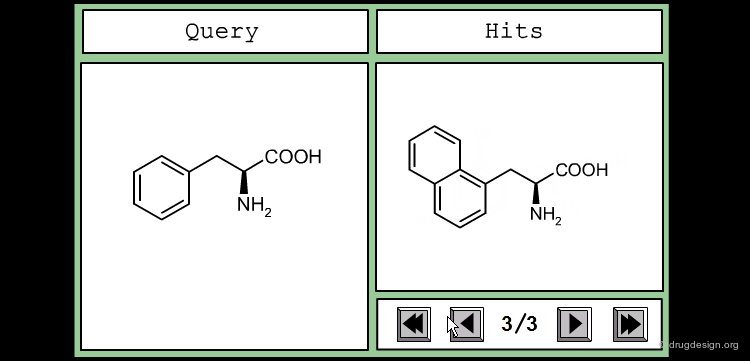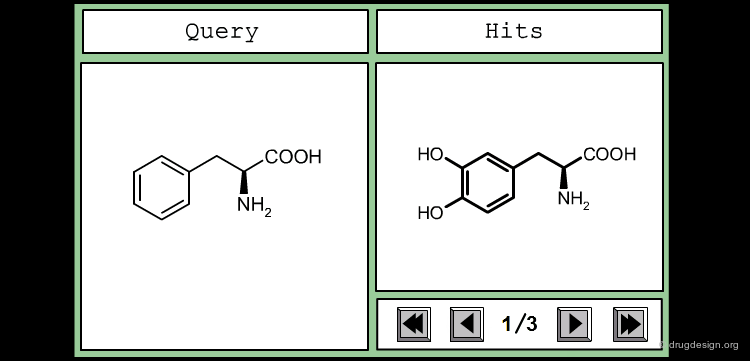
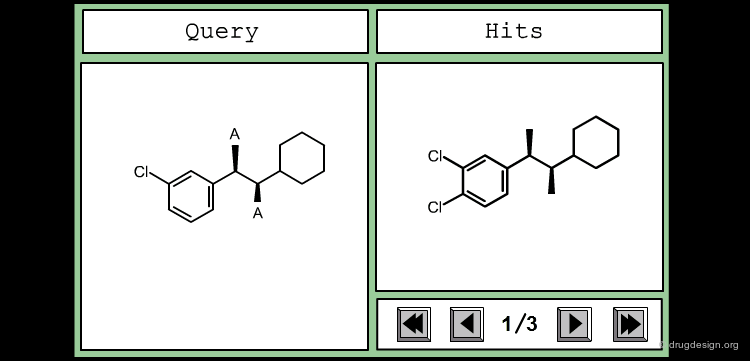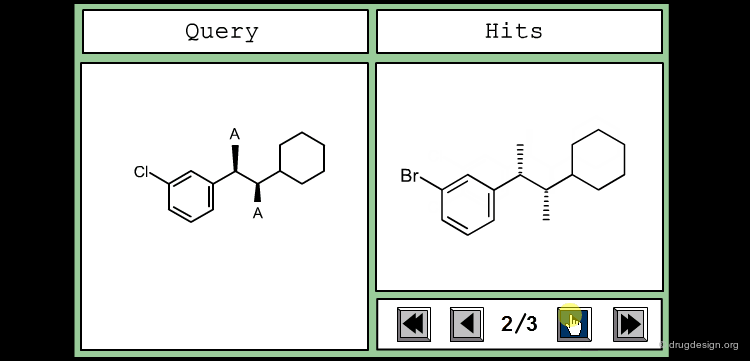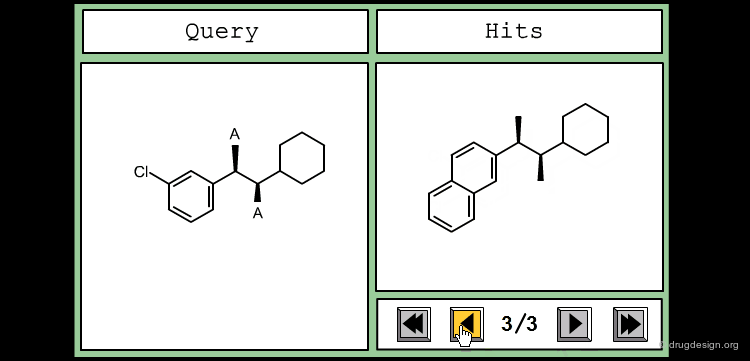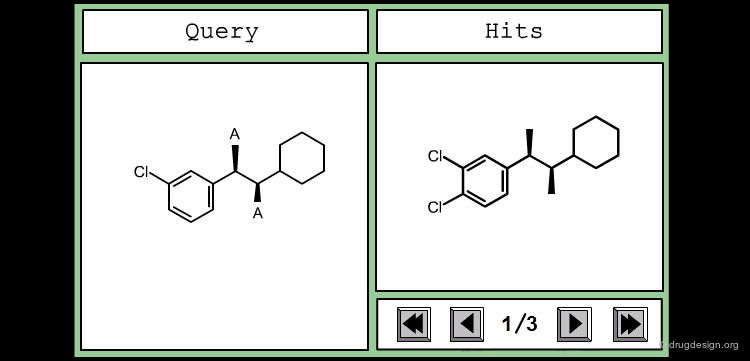

Define Constraints for Tautomers¶
It is possible to prepare the query input in such a way that the hits retrieve tautomers of the target structure according to the constraints defined for this query. The idea is to enable some atoms and bonds to be variable in the query: single or double for the bonds; sp2 or sp3 for the atoms.

Define 3D Constraints¶
It is possible to search in 3D databases for molecules satisfying to precise 3D requirements in the same manner that 2D constraints are introduced when searching 2D databases. This topic is presented in some detail in the chapter entitled "3D Database Searching".

Similarity Search¶
The quest for new ideas can exploit similarity searching with the implicit assumption that similar molecules tend to behave similarly. In this approach a molecule is introduced, and the information system is used to retrieve molecules that resemble the one used as a reference. The similarity is measured with a similarity coefficient (e.g. Tanimoto).

Structural Keys¶
A similarity search is usually done using a fingerprint that encodes the 2D structure into binary descriptors indicating the presence or absence of certain structural features in a molecule. These fingerprints consist of a series of chemical substructure "keys" which enable structural and sub-structural database searching in a very efficient manner.
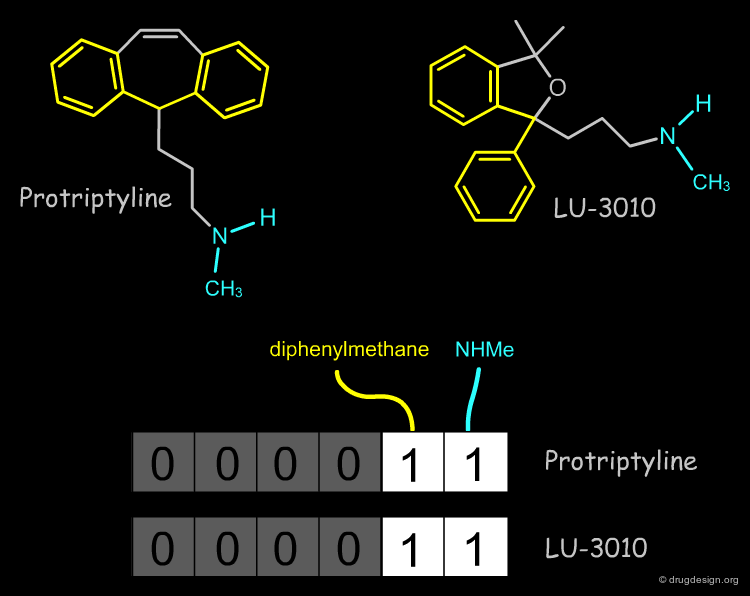
Example of Similarity Measure¶
This is an example of a similarity measure based on the SMILES string. To assess the similarity between the two molecules, we first convert them into their corresponding SMILES representation, which is then used to determine how many substrings of a given length they have in common. We counted all the substrings of length 3 in each molecule, and took the number of common sequences divided by their total number as a measure of similarity.

Similar Name¶
The similarity principle can be extended to search for chemicals having similar names. For example by entering the best guess we have for a chemical name, the activation of the "fuzzy" functionality of the information system will generate chemicals having names that are "similar" to that search name.

Media
Snapshot from ChemDB ChemDB
Focused and Diverse Approaches¶
Constrained and similarity searching can be compared to the "focused" and "diverse" approaches exploited in library design. In general, similarity searching is used when not much information is available (e.g. only one active molecule is known). By contrast, when knowledge has accumulated, a constrained search should be used. The priority is to generate knowledge with a high informational content in an iterative and convergent manner.

Maximizing Knowledge with Information Systems¶
The immediate accessibility of the molecules present in the databases has made the use of information systems a method of choice for knowledge generation. In the example below the information system enables us to explore the pharmacophore responsible for the activity of the benzamide analog. This type of decomposition is sometimes called superstructure search.

Filtering Results¶
Results returned from a search are likely to contain hundred of hits. It is therefore necessary to further narrow down the results and use filters. Filters can be based on structural or property criteria such as logP, molecular weight, number of rotatable bonds, number of hydrogen-bond donors/acceptors, polar surface area etc...
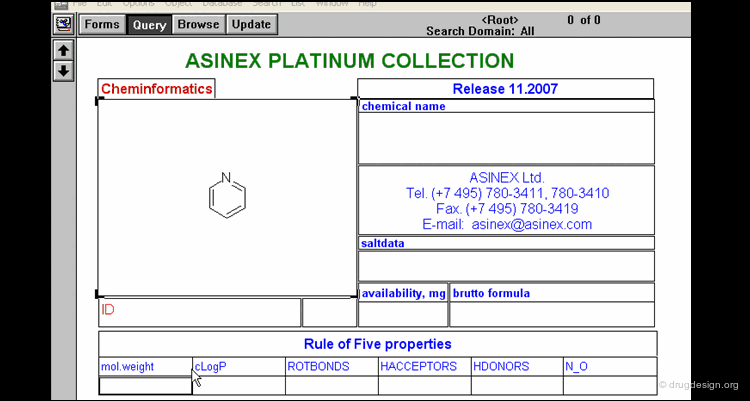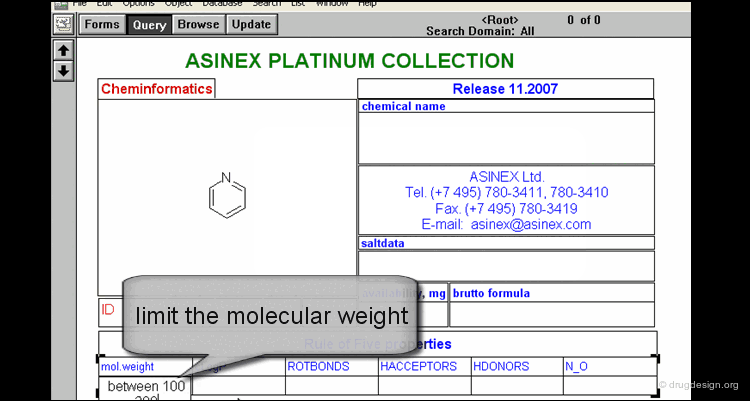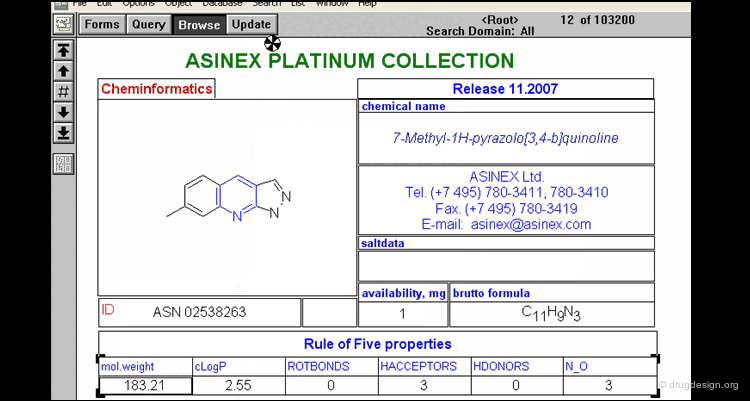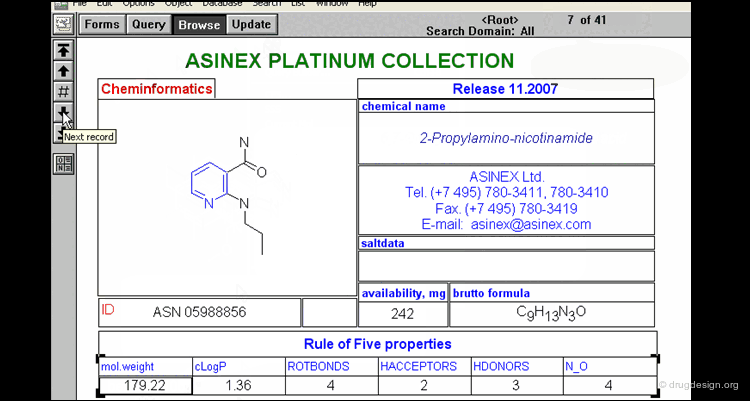
Media
ISIS-Base with ASINEX Database Symyx (MDL)
Boolean Operations with Different Sets of Hits¶
Boolean operations can be used to combine the results of several searches. Suppose a chemist enters a new project where the new target is known to bind to ATP. A useful search could be to find all the molecules in the corporate database that have been synthesized in all ATP binding proteins projects (e.g. kinases, ATP dependent DNA ligases and chaperonins cpn60). The resulting set could then be confronted with a database of toxicophores, to exclude all hits that contain toxic fragments.

Reaction Searching¶
A chemical reaction is "a process that results in the interconversion of chemical species" (IUPAC). Within the context of electronic reaction databases, this general definition of a reaction is limited to the conversion of a reactant set into a product set and can be summed up in the following types of information below :

Author
Gilles Niel Charge de Recherches au CNRS and Professor, Ecole Nationale Superieure de Chimie, Montpellier,France
articles
(IUPAC Recommendations 1994) Glossary of terms used in physical organic chemistry P. Muller Pure and Applied Chemistry 66 (5) 1994
Exact Search and Substructure Search¶
A reaction search is mainly performed through a structure editor that enables exact vs substructure search. Exact search means that all atoms are substituted by implicit Hydrogen atoms (Eq. 1). Substructure search enables some variations on selected atoms (Eq. 2).

Author
Gilles Niel Charge de Recherches au CNRS and Professor, Ecole Nationale Superieure de Chimie, Montpellier,France
Exact Search vs Substructure Search¶
Reaction Substructure Search (RSS) is a very powerfull tool to reach reaction sets of similar reactivity. Eq. 1 specifies the search of experimental conditions necessary to transform the cyclohexene in the presence of N-bromo-succinimide into 3-bromocyclohexene. Beilstein database gave 7 hits (various solvents, concurrent reactions). Eq. 2 is a broadest query since four atoms are labelled as free sites, 10 hits by querying Beilstein database.

Author
Gilles Niel Charge de Recherches au CNRS and Professor, Ecole Nationale Superieure de Chimie, Montpellier,France
Example of Reaction Searching: Query¶
The query shown below, illustrates a substructure search where aspirin is the reactant and the queried products are alkyl esters of aspirin. In this example, substitutions are allowed on all atoms except on the carboxylic function. Further rings on the benzene ring are not allowed.

Author
Gilles Niel Charge de Recherches au CNRS and Professor, Ecole Nationale Superieure de Chimie, Montpellier,France
Example of of Reaction Searching: Results¶
The resulting answer set contains 4 hits. Only the first one is shown on the screenshot. It is a simple esterification using diazomethane as a reactant. Each reactant or reagent on the reaction scheme is clickable and thus searchable as a substance in any other reaction. A link to the reference article is offered to get further details on the reaction.

Author
Gilles Niel Charge de Recherches au CNRS and Professor, Ecole Nationale Superieure de Chimie, Montpellier,France
Querying Modes¶
Electronic reaction databases enable several querying modes according to the user's needs. Six examples of questions are indicated below. The two first querying modes are probably the most used by organic chemists. Within an answer set other queries may specify the experimental conditions, yield, relevant authors, etc. The reaction selectivity principle is applicable to functional groups as well as to bond changes (make/break, stereochemistry).

Author
Gilles Niel Charge de Recherches au CNRS and Professor, Ecole Nationale Superieure de Chimie, Montpellier,France
Structure Editor¶
A structure editor enables to specify the atom status of reactants, products and dynamic changes that occur during chemical reaction. The main common features of available structure editors are outlined below.

Author
Gilles Niel Charge de Recherches au CNRS and Professor, Ecole Nationale Superieure de Chimie, Montpellier,France
Drawing Atoms¶
The vertical and horizonatal toolbars are extracted from Scifinder which is the required software to query the Chemical Abstracts Service (CAS) database. Here is shown the vertical toolbar that enables drawing atoms, chains, cycles, etc. Some icons are self-expressive and a detailed information is given on the CAS site.

Author
Gilles Niel Charge de Recherches au CNRS and Professor, Ecole Nationale Superieure de Chimie, Montpellier,France
Drawing Bonds¶
Here is shown the vertical toolbar that enables drawing atoms, chains, cycles, etc.

Author
Gilles Niel Charge de Recherches au CNRS and Professor, Ecole Nationale Superieure de Chimie, Montpellier,France
Atom-to-Atom Mapping¶
Atom-to-atom mapping explicitly assigns atoms of the reactant structures to corresponding atoms of the product structures (Gasteiger and Engel, 2003). The mapping is necessary to get relevant hits to a given query. Let's see the example below; the queries 1 and 2 only differ from the presence or the absence of the mapping. The mapping brings the Query 2 to a satisfactory precision level.

Author
Gilles Niel Charge de Recherches au CNRS and Professor, Ecole Nationale Superieure de Chimie, Montpellier,France
book
J. Gasteiger and T. Engel Chemoinformatics, A Textbook Weinheim: Wiley 2003
Control on the Reaction Mechanism¶
The given answer to Query 1 (no mapping) is chemically correct (methylation alpha to a ketone) but is unexpected since both the ketone and the dioxolane ring remain unchanged during the reaction; Query 1 is not well-defined enough. Mapping a reaction query enables the user to exert a more accurate control on the reaction mechanism.

Author
Gilles Niel Charge de Recherches au CNRS and Professor, Ecole Nationale Superieure de Chimie, Montpellier,France
Definitions for Most Reaction Database Editors¶
Some fundamental definitions are common to most reaction database editors as follows: (1) a reactant contributes at least one carbon atom to a reaction product, and may also contribute noncarbon atoms; (2) a reagent can contribute only noncarbon atoms to a reaction product; (3) a catalyst initiates or promotes the action of other participants in a reaction.

Author
Gilles Niel Charge de Recherches au CNRS and Professor, Ecole Nationale Superieure de Chimie, Montpellier,France
Specifying Reagents¶
Depending on the reaction database editor, specifying the reagent nature will take a different course. Using Scifinder, reagents and reactants are drawn as reactants in a chemical reaction though they may be assigned a unique role if they are queried as substances only. Beilstein and MDL/ISIS databases offer specific querying tools for both reagent vs reactant.

Author
Gilles Niel Charge de Recherches au CNRS and Professor, Ecole Nationale Superieure de Chimie, Montpellier,France
Chemical Reaction Databases¶
An overview of the main chemical reaction databases is given in the table below. The number of reactions correspond to the January 2008 values; these databases are continually updated.

Author
Gilles Niel Charge de Recherches au CNRS and Professor, Ecole Nationale Superieure de Chimie, Montpellier,France
book
Methods of Organic Chemistry Thieme publisher 2001
Science of Synthesis: Houben-Weyl Methods of Molecular Transformations Georg Thieme Verlag, Stuttgart 2005
Organic Reactions Jossey-Bass publ. 61 volumes
Comprehensive Organic Chemistry Elsevier 1979
Comprehensive Organic Functional Group Transformations Elsevier 1995
Comprehensive Heterocyclic Chemistry I and II Pergamon Press 1984 - 1996
Comprehensive Natural Products Chemistry Pergamon Press 1999
Organic Syntheses
1921-2005
Encyclopedia of Reagents for Organic Syntheses Wiley 1995 +
Reagents for Organic Synthesis
1967 +
Handbook of Reagents for Organic Syntheses Wiley 2000
Comprehensive Organic Transformations Wiley 1999
The Chemistry of Functional Groups Wiley Guide 1992
Compendium of Organic Synthetic Methods Wiley
Advanced Organic Chemistry Jossey-Bass 2001
Synthetic Methods of Organic Chemistry Karger Publ.
Comprehensive Organic Synthesis Pergamon Press 1991 +
The Logic of Chemical Synthesis Wiley 1989
Protective Groups in Organic Synthesis Wiley 1999
Contemporary Organic Synthesis The Royal Society of Chemistry 1994 +
Comparison CASREACT vs Beilstein¶
As seen on the previous slide, CASREACT and Beilstein are the most important reaction databases. However they are somewhat different.

Author
Gilles Niel Charge de Recherches au CNRS and Professor, Ecole Nationale Superieure de Chimie, Montpellier,France
Multistep Reactions¶
The CAS indexation policy in case of multi-step reactions (A -> B -> C -> D) is to index every single step AND every intermediate step (A -> C, B -> D, A -> D). As a consequence the total number of reactions is dramatically increased and less than half of reactions are single step reactions. Beilstein reaction database indexes mainly (98%) single step reactions.

Author
Gilles Niel Charge de Recherches au CNRS and Professor, Ecole Nationale Superieure de Chimie, Montpellier,France
Indexes¶
CASREACT indexes only full reactions while about 25 % reactions in Beilstein are "half-reactions" defined by the fact, that either only reactants or only products are characterized by a Beilstein Registry Number.

Author
Gilles Niel Charge de Recherches au CNRS and Professor, Ecole Nationale Superieure de Chimie, Montpellier,France
Complementarity¶
The two databases are complementary since the Beilstein database has indexed substances and their associated reactions a long time before the CAS and CASREACT (1779 vs 1956).

Author
Gilles Niel Charge de Recherches au CNRS and Professor, Ecole Nationale Superieure de Chimie, Montpellier,France
Polymers, Peptides and Nucleotides¶
In addition reactions about polymers, peptides or nucleotides are only searchable in CAS databases since these substances are not indexed in Beilstein.

Author
Gilles Niel Charge de Recherches au CNRS and Professor, Ecole Nationale Superieure de Chimie, Montpellier,France
Management of Reaction Lists¶
Reaction lists can be managed in terms of union, intersection or exclusion in both databases. Neverteheless Beilstein offers an interesting clusterizing tool to categorize a reaction set based on the similarity level of the reaction center (also present in ChemInform but not in CAS).

Author
Gilles Niel Charge de Recherches au CNRS and Professor, Ecole Nationale Superieure de Chimie, Montpellier,France
ChemInform¶
This reaction database is the most important reaction database designed by FIZ Chemie and edited today under MDL/ISIS technology. The journal coverage is similar to that of CAS and Beilstein, the difference on the number of reactions comes from the data selectivity. Only new synthetic methodologies are indexed. Searching within this database is thus oriented towards the synthetic method retrieval. Tools for drawing and specifying a reaction scheme are especially well-designed in ChemInform. Many clusterizing features are allowed including reagent type, reaction classification based on the reaction center similarity etc.

Author
Gilles Niel Charge de Recherches au CNRS and Professor, Ecole Nationale Superieure de Chimie, Montpellier,France
Data Analysis¶
Introduction to QSAR Modeling¶
Quantitative Structure-Activity Relationships (QSAR) and Quantitative Structure-Property Relationships (QSPR) are based on the fundamental assumptions of Corwin Hansch (1964), who is considered to be the father of QSAR: "The molecular structure of a chemical influences its biological activity and physical-chemical properties; similar compounds behave similarly".

Author
Paola Gramatica QSAR Research Unit in Environmental Chemistry and Ecotoxicology, DBSF - University of Insubria, Varese, Italy
wikipedia
QSAR Definition¶
Finding and understanding a chemical's structural characteristics related to a particular property or activity allows for the development of a mathematical function f to correlate to the chemical's structure and behavior. This function f can be used in a mathematical formula to predict data for compounds that have not been tested, or even not yet synthesized. QSAR studies organize the existing knowledge of an endpoint (activity or property) with the purpose of generalizing such knowledge. This allows predictions to be made for other chemicals without available data.

Author
Paola Gramatica QSAR Research Unit in Environmental Chemistry and Ecotoxicology, DBSF - University of Insubria, Varese, Italy
articles
The Use Of Substituent Constants In The Analysis Of The Structure--Activity Relationship In Penicillin Derivatives. Hansch C, Steward Ar. J Med Chem. 7 1964
book
Corwin Hansch and Albert Leo Substituent constants for correlation analysis in chemistry and biology. Wiley, 605 Third Ave., New York, 1979
The QSPR/QSAR Problem¶
The molecular structure of an organic compound determines the properties of the chemical. An indirect approach must be used which consists of two main parts: (a) each compound's molecular structure is represented by calculating molecular descriptors and (b) subsets of the descriptors are chosen and good models predicting the property or activity of interest are built. The method is inductive, as it depends on having a set of compounds with experimentally known activities or properties. This set of known compounds is used to develop the model further.

Author
Paola Gramatica QSAR Research Unit in Environmental Chemistry and Ecotoxicology, DBSF - University of Insubria, Varese, Italy
SAR Definition¶
Structure-Activity Relationships (SAR) studies are based on the identification of structural fragments (alerts) related to a particular activity, through Similarity Analysis and Expert Systems methods. An SAR is a qualitative relationship (an association) between a molecular (sub)structure and the presence or absence of an activity. Some historical and pioneering SARs are listed below.
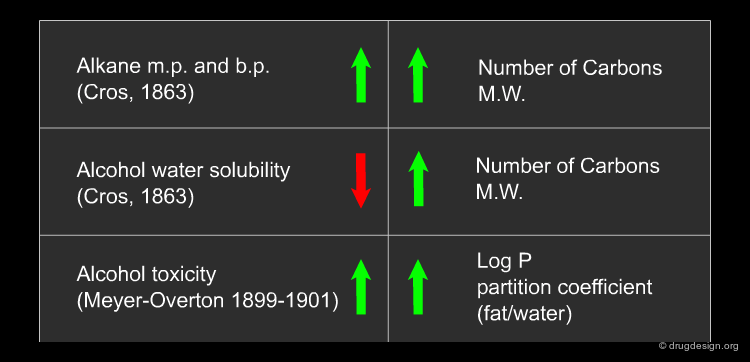
Author
Paola Gramatica QSAR Research Unit in Environmental Chemistry and Ecotoxicology, DBSF - University of Insubria, Varese, Italy
Qualitative Class Assignment of New Chemicals¶
Substructures associated with the presence of biological activity are sometimes called biophores, whereas those associated with the absence of activity are called biophobes. Descriptors, selected from a "learning set" of active and inactive molecules, allow for a qualitative class assignment of new chemicals, containing activating descriptors (biophores) or inactivating descriptors (biophobes).
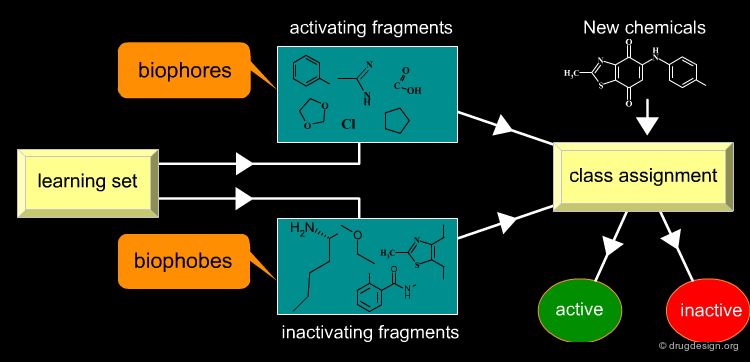
Author
Paola Gramatica QSAR Research Unit in Environmental Chemistry and Ecotoxicology, DBSF - University of Insubria, Varese, Italy
Three Prerequisites for QSAR Modeling¶
The three prerequisites for QSAR modeling are: (1) an experimental data set: a 'limited' number of experimental input data, on which to find the Structure-Activity Relation and to develop QSARs. They must be as numerous as possible, correct, representative and homogenous (same lab, ideally same researcher). The models will only be as good as the data used to develop them: 'Garbage in, garbage out' (2) molecular descriptors: used to translate the chemical structure features into numbers and (3) statistical methods: applied to develop quantitative models between a response, the dependent variable (Y), and one or more molecular descriptors, namely independent variables (X). The models must have validated predictive performances.

Author
Paola Gramatica QSAR Research Unit in Environmental Chemistry and Ecotoxicology, DBSF - University of Insubria, Varese, Italy
Classical Hansch Equation¶
The classical Hansch equation is a Multiple Linear Regression Model (MLR) relating a studied biological activity to a combination of different molecular properties/descriptors selected by the modeler as informative of specific chemical behavior. Hansch models are mainly applicable to congeneric chemicals, in which a substitution variation of a common basic structure occurs.

Author
Paola Gramatica QSAR Research Unit in Environmental Chemistry and Ecotoxicology, DBSF - University of Insubria, Varese, Italy
wikipedia
Molecular Descriptors Calculations¶
Different molecular descriptors are different ways or perspectives for viewing a chemical. The molecular structure of each compound is entered and stored in a topological representation. Each structure is submitted to conformational analysis to generate a good, lower energy conformation. The topological and geometrical representation of the structures, which give the atomic x,y,z coordinates, are used to calculate molecular structure descriptors. Various software packages are available for molecular descriptor calculation such as ADAPT, OASIS, CODESSA, MolConnZ, DRAGON and MOPAC.
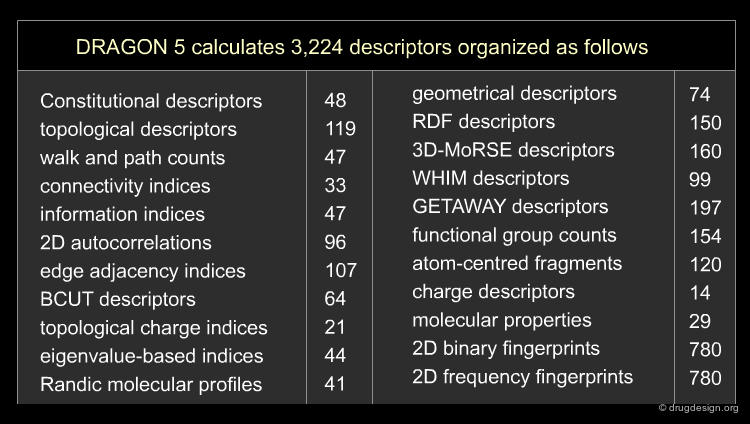
Author
Paola Gramatica QSAR Research Unit in Environmental Chemistry and Ecotoxicology, DBSF - University of Insubria, Varese, Italy
articles
OASIS method for predicting biological activity of chemical compounds Mekenyan O, Bonchev D. Acta Pharm Jugosl 36 1986
The OASIS Concept for Predicting the Biological Activity of Chemical Compounds O. Mekenyan, S. Karabunarliev and D. Bonchev, J. Math. Chem. 4 1990 10.1007/BF01170013
CODESSA Reference Manual (version 2.0), Gainesville, FA A.R. Katritzky, V.S. Lobanov AND M. Karelson
1994
book
L.H. Hall
Hall Associates Consulting; Eastern Nazaree College, Quincy, Massachusetts 02170, USA. 2003
Roberto Todeschini and Viviana Consonni Methods and Principles in Medicinal Chemistry Volume 11 Wiley-VCH 2000
Theoretical Molecular Descriptors¶
Molecular descriptors translate chemical structure features into numbers. These are different perspectives or ways to view a molecule: mono-dimensional (1D) such as atom or group counts, bi-dimensional (2D) such as topological or connectivity from the molecular graph, or three-dimensional (3D) from a minimum energy conformation. Other properties (as logP) and quantum chemical descriptors (such as HOMO, LUMO, etc) are also widely used in QSAR models.
Author
Paola Gramatica QSAR Research Unit in Environmental Chemistry and Ecotoxicology, DBSF - University of Insubria, Varese, Italy
Chemometric Approaches to QSAR Modeling¶
Chemicals with available experimental data (Y) and calculated molecular descriptors (X) are used as training sets on which to find the QSAR. Explorative methods (such as PCA or Cluster Analysis) give the researcher a 'view' of the chemical domain, highlighting compounds that are too peculiar (possible structural outliers) which could be excluded from the training set.

Author
Paola Gramatica QSAR Research Unit in Environmental Chemistry and Ecotoxicology, DBSF - University of Insubria, Varese, Italy
Development of Quantitative Models¶
Regression methods (as MLR, PLS, etc.) develop quantitative models for quantitative responses (a potency), while classification methods (such as CART, DA, Neural Networks, etc.) develop quantitative models for qualitative responses (a category).

Author
Paola Gramatica QSAR Research Unit in Environmental Chemistry and Ecotoxicology, DBSF - University of Insubria, Varese, Italy
Identify the Best Subset of Descriptors¶
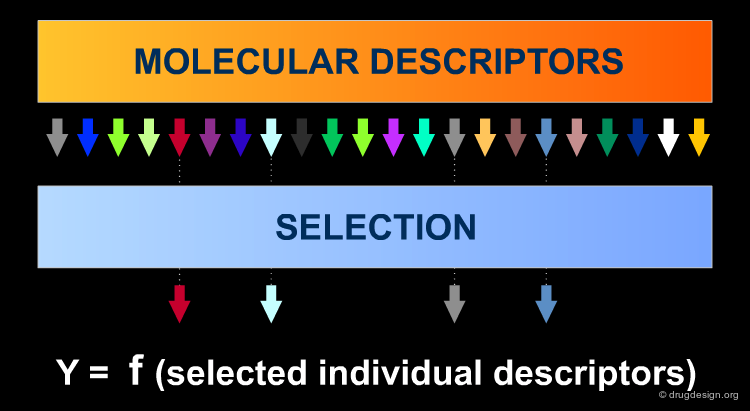
The set of calculated descriptors must be reduced to a set of descriptors which is informationally rich but as small as possible (Ockham's Razor: "avoid complexity if not necessary"). The relevant Variable Selection (VS) can be made subjectively by the modeler based on experience, tradition, availability, or, better still, performed mathematically. The identification of the best subset of descriptors is made in two steps.
Author
Paola Gramatica QSAR Research Unit in Environmental Chemistry and Ecotoxicology, DBSF - University of Insubria, Varese, Italy
Variable Selection: Independent Variables X¶
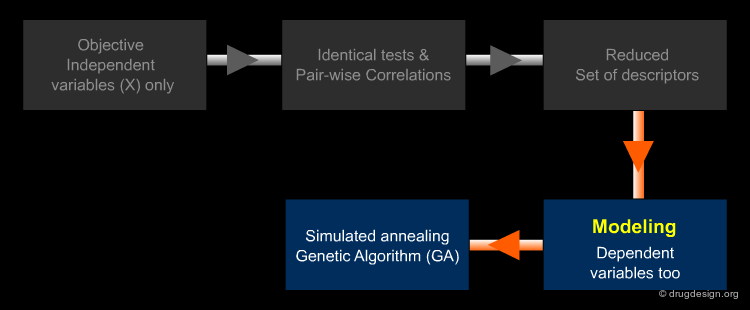
For the first step a large number of molecular descriptors can be calculated as input, in order to have exhaustive information a-priori about different structural features since a molecule is a complex system which can be 'viewed' in different ways. Objective selection uses only independent variables X (descriptors). Descriptors to discard are identified by tests of identical values and pairwise correlations, by looking for descriptors orthogonal to one another.

Author
Paola Gramatica QSAR Research Unit in Environmental Chemistry and Ecotoxicology, DBSF - University of Insubria, Varese, Italy
Variable Selection: Dependent Variables Y¶
Secondly, only relevant information really related to the target response must be identified: this is the role of modeling variable selection methods (such as Genetic Algorithms). Modeling VS, which also uses dependent variable values (Y), is applied to further reduce the descriptor set to the true modeling set.
Author
Paola Gramatica QSAR Research Unit in Environmental Chemistry and Ecotoxicology, DBSF - University of Insubria, Varese, Italy
Characteristics of QSAR Models¶
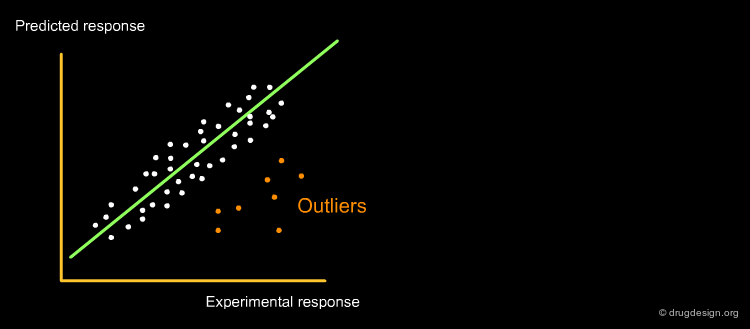
QSAR models must be verified for their statistical qualities, in fitting performance by the determination coefficient R2, and in prediction power: both internal by Cross-Validation (R2cv or Q2LOO/LMO) and external by Q2EXT or R2EXT. The response outliers, i.e. poorly predicted chemicals, must also be highlighted. The reliability of the predicted data must be verified by defining the applicability domain (AD), namely the model descriptor space, because only predictions for chemicals in the model AD can be considered as not extrapolated. A classical plot for a QSAR model is shown below.

Author
Paola Gramatica QSAR Research Unit in Environmental Chemistry and Ecotoxicology, DBSF - University of Insubria, Varese, Italy
Example of QSAR Model¶
Below is shown an example of a QSAR model for acute toxicity in Pimephales promelas, based on six theoretical molecular descriptors. The information related to molecular size is mainly condensed in WA and Mv, the information related to the electronic distribution is represented by MAXDP. Other counters (nN, nCb, and H-046) are needed to model some specific chemicals in the dataset.



Author
Paola Gramatica QSAR Research Unit in Environmental Chemistry and Ecotoxicology, DBSF - University of Insubria, Varese, Italy
articles
Statistically validated QSARs and theoretical descriptors for the modelling of the aquatic toxicity of organic chemicals in Pimephales promelas (Fathead Minnow) Ester Papa, Fulvio Villa, and Paola Gramatica J.Chem.Inf.Model. 45 2005 10.1021/ci050212l
wikipedia
Chemical Domain of Applicability¶
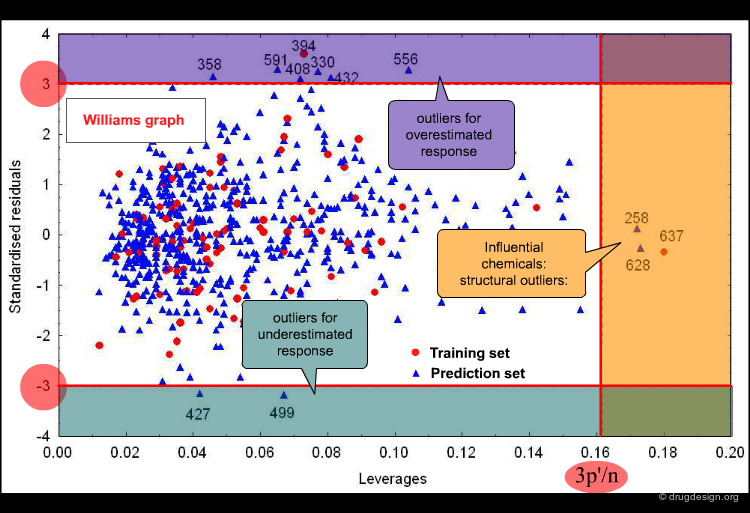
The applicability domain (AD) of a QSAR model can be verified by different tools. One, which is widely used in regression models, is based on the chemical distance from the model space. The Williams plot of cross-validated standardized residuals vs. leverages (Hat diagonal) values allows for simple graphic detection of both the response outliers (Y outliers) and the structurally influential chemicals (X outliers) in a model.

Author
Paola Gramatica QSAR Research Unit in Environmental Chemistry and Ecotoxicology, DBSF - University of Insubria, Varese, Italy
articles
Current Status of Methods for Defining the Applicability Domain of (Quantitative) Structure-Activity Relationships Netzeva, T. I., Worth, A. P., Aldenberg, T., Benigni, R., Cronin, M. T. D., Gramatica, P., Jaworska, J. S., Kahn, S., Klopman, G., Marchant, C. A., Myatt, G., Nikolova-Jeliazkova, N., Patlewicz, G. Y., Perkins, R., Roberts, D. W., Schultz, T. W., Stanton, D. T., Sandt, J. J. M. van de, Tong, W. D., Veith, G., Yang, C. H. ATLA, Alternatives to Laboratory Animals 33 (2) 2005
Methods for reliability, uncertainty assessment, and applicability evaluations of regression based and classification QSARs Lennart Eriksson, Joanna Jaworska, Andrew Worth, Mark Cronin, Robert M McDowell, Paola Gramatica Environ. Health Perspectives 111 (10) 2003 10.1289/ehp.5758
Application Domain from Williams plot¶
The horizontal lines indicate the limits for normal values: compounds with residuals +/-3 Σ are Y outliers. The limit for normal values of the X outliers (vertical line) is calculated by 3 p'/n (p' = number of model variables + 1, n = number of objects used to calculate the model).
Author
Paola Gramatica QSAR Research Unit in Environmental Chemistry and Ecotoxicology, DBSF - University of Insubria, Varese, Italy
Validity Check and Predictivity¶
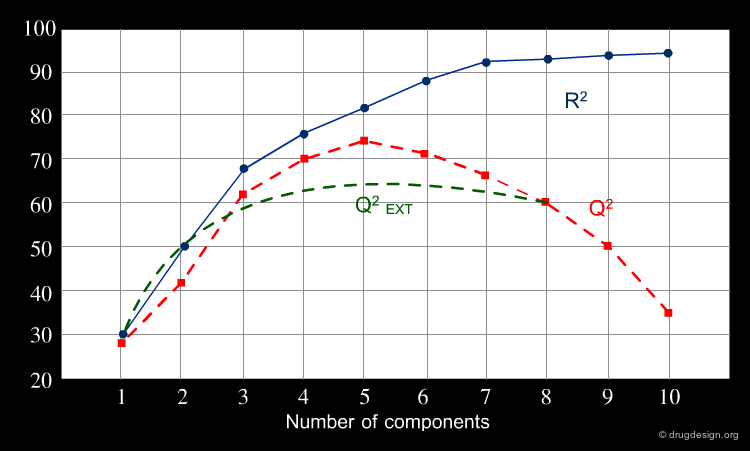
A model simply explaining the known data (fitting model) is not sufficient, if reliable predicted data are needed. Not all models with good fitting performances (high R2) are predictive (high Q2 or R2 cv), particularly for new chemicals (high Q2 EXT). A validity check of the developed QSAR model for the prediction of new compounds is necessary. The graph shows the plot of a frequent situation: the fitting increases with increasing descriptor numbers, while predictivity, generally worse, can dramatically decline. Only externally validated models can provide reliable predictions.
Author
Paola Gramatica QSAR Research Unit in Environmental Chemistry and Ecotoxicology, DBSF - University of Insubria, Varese, Italy
articles
The importance of being Earnest: Validation is the absolute essential for successful application and interpretation of QSPR models A.Tropsha, P. Gramatica, V.K. Gombar QSAR and Comb. Sci. 22 2003 10.1002/qsar.200390007
Validation Parameters of QSAR Models¶
To ensure reliable predicted data, a model's predictive performance must be verified by statistical parameters. For regression models, the most commonly used parameters are shown below (click the different parameters to get more details).

Author
Paola Gramatica QSAR Research Unit in Environmental Chemistry and Ecotoxicology, DBSF - University of Insubria, Varese, Italy
articles
Principles of QSAR models validation: internal and external P. Gramatica QSAR and Comb.Sci. 26 2007 10.1002/qsar.200610151
wikipedia
Statistic of QSAR Classification Models¶
Classification, also called supervised pattern recognition, is aimed at developing a classification rule, i.e. a quantitative model (selecting predictor variables: the molecular descriptors) based on a training set of objects of known classes (the qualitative responses) so that the rule can be applied to a test set of objects of unknown classes.

Author
Paola Gramatica QSAR Research Unit in Environmental Chemistry and Ecotoxicology, DBSF - University of Insubria, Varese, Italy
Classification Methods¶
There is a wide range of classification methods, including: Discriminant Analysis (DA), SIMCA (Soft Independent Modeling of Class Analogy), kNN (k Nearest Neighbors), CART (Classification And Regression Tree) etc. For a classic two group classification (active/inactive) the following statistic can be applied (click the different items to get more details).

Author
Paola Gramatica QSAR Research Unit in Environmental Chemistry and Ecotoxicology, DBSF - University of Insubria, Varese, Italy
wikipedia
Scheme for Predictive QSAR Modeling¶
In the following scheme the approach for predictive QSAR modeling is depicted. If no new experimental data are available, the available dataset can be split, before the modeling, by various methods (similarity distance-based, as k-NN, Self Organizing Maps (SOM), D-optimal design, random etc.) This will yield an external prediction set of chemicals, that are not involved in model development on which to verify the quality of the fitted and cross-validated model. Only externally predicted models have sufficient and verified generalizability for the production of reliable, new predicted data.

Author
Paola Gramatica QSAR Research Unit in Environmental Chemistry and Ecotoxicology, DBSF - University of Insubria, Varese, Italy
articles
Principles of QSAR models validation: internal and external P. Gramatica QSAR and Comb.Sci. 26 2007 10.1002/qsar.200610151
The importance of being Earnest: Validation is the absolute essential for successful application and interpretation of QSPR models A.Tropsha, P. Gramatica, V.K. Gombar QSAR and Comb. Sci. 22 2003 10.1002/qsar.200390007
Reversible Decoding of Molecular Descriptors¶
A predictive QSAR model can be applied to new chemicals, also those not yet synthesized, for new predicted data. If the selected molecular descriptors are also interpreted for mechanistic meaning (descriptive QSAR), their reversible decoding is the best basis for chemical design.

Author
Paola Gramatica QSAR Research Unit in Environmental Chemistry and Ecotoxicology, DBSF - University of Insubria, Varese, Italy
Interpretation of Molecular Descriptors¶
The interpretation of the selected descriptors, which provides their mechanistic meaning in relation to the modeled response, is particularly important when the aim of the QSAR modeling is information on the mechanism, namely descriptive QSAR. However descriptors selected by variable selection methods as "best correlated to response" are not necessarily the best for an easy understanding of the complexity of the target response.

Author
Paola Gramatica QSAR Research Unit in Environmental Chemistry and Ecotoxicology, DBSF - University of Insubria, Varese, Italy
articles
The characterization of chemical structures using molecular properties. A survey. Livingstone, D.J. J. Chem. Inf. Comput. Sci. 40 2000 10.1021/ci990162i
Predictive and Descriptive QSAR Models¶
If the molecular descriptors are not always plainly interpretable, their practical value relies mainly on their predictive ability, which must be carefully validated: this is the role of predictive QSAR. If reliable predicted data are needed, then "a validated mathematical model relating a target property to chemical features may, in some cases, be all that is necessary" (Livingstone).

Author
Paola Gramatica QSAR Research Unit in Environmental Chemistry and Ecotoxicology, DBSF - University of Insubria, Varese, Italy
articles
QSAR for Boiling Points of "Small" Sulfides. Are the "High-Quality Structure-Property-Activity Regressions" the Real High Quality QSAR Models? Zefirov N. S. and Palyulin V. A. J. Chem. Inf. Comput. Sci 41 2001 10.1021/ci0001637
The characterization of chemical structures using molecular properties. A survey. Livingstone, D.J. J. Chem. Inf. Comput. Sci. 40 2000 10.1021/ci990162i
OECD Principles for QSAR Models¶
In 2004, OECD Principles were defined for the validation, for regulatory purposes, of (Q)SAR models. The new European regulation, in which the following principles will be applied, is called REACH: Regulation, Evaluation and Authorization of Chemicals. To facilitate examination of a QSAR model for regulatory purposes, it should be associated with the following information.

Author
Paola Gramatica QSAR Research Unit in Environmental Chemistry and Ecotoxicology, DBSF - University of Insubria, Varese, Italy
Main Applications of QSAR Predictions¶
The main applications of QSAR predictions are: filling of data gaps, validation of experimental data, screening and ranking, chemical design and highlighting chemicals of interest (also before their synthesis).
Author
Paola Gramatica QSAR Research Unit in Environmental Chemistry and Ecotoxicology, DBSF - University of Insubria, Varese, Italy
Applications of Cheminformatics¶
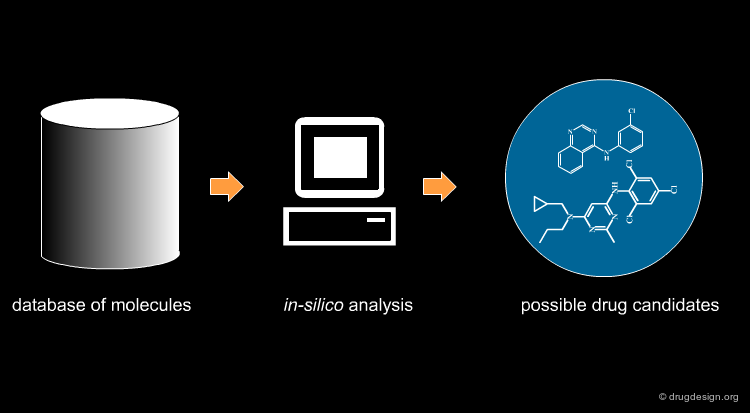
Virtual Screening¶
Virtual screening (VS) is a computational approach used in drug discovery. It is based on a rapid in silico analysis of large databases of chemical compounds in order to identify possible drug candidates. VS is also called "virtual high throughput screening", or "in-silico screening".
Author
Darren Fayne et al. Senior Research Fellow, Molecular Design Group, School of Biochemistry and Immunology, Trinity College Dublin, Ireland
Use of Virtual Screening in Research¶
Virtual screening is used to select or prioritize compounds for experimental screening. The molecules are obtained either from a pool of available compounds, or by chemical syntheses. In both cases it helps to reduce the number of compounds to be ordered or synthesized to a manageable size, and to be compatible with the capabilities in chemistry and in the biological test.

Author
Darren Fayne et al. Senior Research Fellow, Molecular Design Group, School of Biochemistry and Immunology, Trinity College Dublin, Ireland
VS, an Essential Tool to Library Design¶
The introduction of combinatorial chemistry in the mid eighties has provided to the medicinal chemists powerfull means to synthesize large libraries of molecules. The last few years have seen a shift in the strategy where pharmaceutical companies try to reduce their costs by concentrating in the design of well conceived focused libraries. Virtual Screening is the right tool for that.

Author
Darren Fayne et al. Senior Research Fellow, Molecular Design Group, School of Biochemistry and Immunology, Trinity College Dublin, Ireland
Virtual Screening Guides Compound Exploration¶
Virtual screening can be used for compound exploration. The molecules generated by virtual screening can be represented in the space of their properties and compared with different groups of known active molecules. Four different classes of biological activities are illustrated here.

Author
Darren Fayne et al. Senior Research Fellow, Molecular Design Group, School of Biochemistry and Immunology, Trinity College Dublin, Ireland
Ligand-Based and Structure-Based VS¶
Virtual screening methods can be either ligand-based or structure-based. In the first case the focus is on the selection of molecules having similarities with a given reference structure; in the second case the 3D structure of the target is exploited to identify those molecules exhibiting the most favorable interactions.

Author
Darren Fayne et al. Senior Research Fellow, Molecular Design Group, School of Biochemistry and Immunology, Trinity College Dublin, Ireland
Ligand-Based Methods¶
The "molecular similarity principle", is the underlying concept of all ligand-based VS methods. It states that similar molecules tend to behave similarly, while more dissimilar molecules exhibit more distinct properties. Typically, the similarity is assessed by considering two- or three-dimensional chemistry, shape, electrostatic, and interaction points (e.g., pharmacophore points). All ligand-based VS methods have in common the goal of identifying similar compounds (see figure below).

Author
Darren Fayne et al. Senior Research Fellow, Molecular Design Group, School of Biochemistry and Immunology, Trinity College Dublin, Ireland
Darren Fayne et al. Senior Research Fellow, Molecular Design Group, School of Biochemistry and Immunology, Trinity College Dublin, Ireland
2D Substructure Search¶
2D substructure searches can be used to find molecules that are similar to a reference compound. Key structural moieties of the reference compound are identified and used as a query to search for hits in a database of molecules.

Author
Darren Fayne et al. Senior Research Fellow, Molecular Design Group, School of Biochemistry and Immunology, Trinity College Dublin, Ireland
2D Fingerprint Similarity¶
An alternative to 2D structural search are methods based on 2D fingerprints. In this case it is not necessary to define a query; the whole molecule is automaticaly scanned for the presence or absence of thousands of structural features (binary descriptors), forming the fingerprint of the reference molecule. The VS software will search in the database for molecules with similar fingerprints.
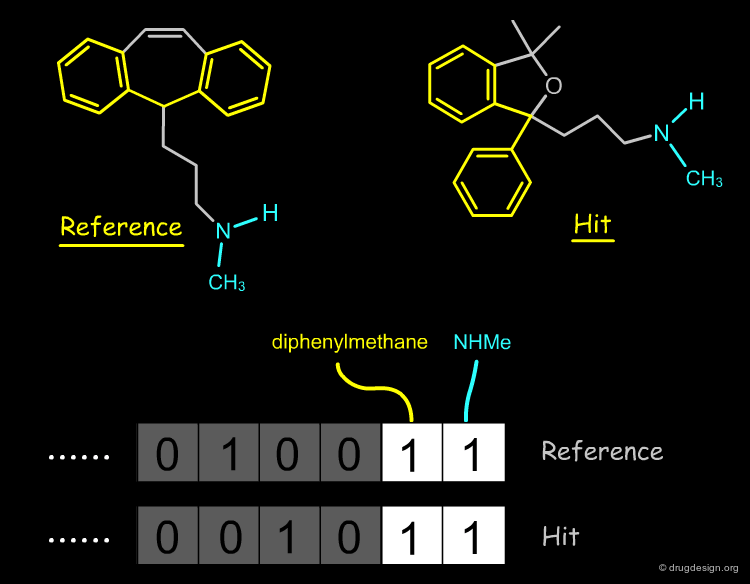
Author
Darren Fayne et al. Senior Research Fellow, Molecular Design Group, School of Biochemistry and Immunology, Trinity College Dublin, Ireland
3D Pharmacophore¶
An extension of 2D sub-structure searches is 3D pharmacophore searches. Instead of defining 2D key structural elements in the query, an abstract pharmacophore is used, which represents a specific 3D arrangement of chemical groups assumed to be essential for the biological activity. The VS program will select in the database molecules with similiar pharmacophoric arrangments. This method is highly dependent on ligand conformations and on the consideration of conformational multiplicity for the ligand.


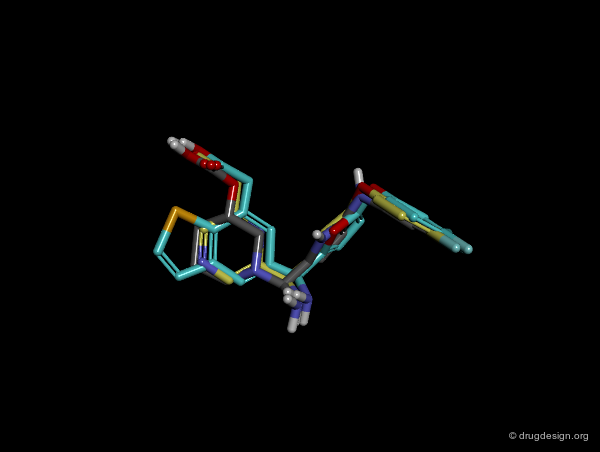
Author
Darren Fayne et al. Senior Research Fellow, Molecular Design Group, School of Biochemistry and Immunology, Trinity College Dublin, Ireland
Ligand Shape Similarity¶
When VS is based on shape similarity, the molecules of a database that possess a given shape are candidate compounds that can be considered for experimental screening.

Author
Darren Fayne et al. Senior Research Fellow, Molecular Design Group, School of Biochemistry and Immunology, Trinity College Dublin, Ireland
QSAR¶
QSAR tackles the issue of compound selection more mathematically. It tries to find a mathematical equation which correlates the properties of a set of reference structures with their biological activities. VS methods based on QSAR use this mathematical model to scan relevant molecules of a database and predict their activities.

Author
Darren Fayne et al. Senior Research Fellow, Molecular Design Group, School of Biochemistry and Immunology, Trinity College Dublin, Ireland
MIF Analysis¶
When VS is based on MIF (Molecular Interaction Field) similarity, the molecules of a database that possess similar MIFs are candidate compounds that can be considered for experimental screening. This is an important component of 3D-QSAR methods.

Author
Darren Fayne et al. Senior Research Fellow, Molecular Design Group, School of Biochemistry and Immunology, Trinity College Dublin, Ireland
Property-Based Filtering of the VS Results¶
The principle of property-based filtering of the VS results is the following: the physical and chemical properties of each hit are calculated. These are then compared to molecules with the desired physiological characteristics. The selection of new molecules for wet-lab evaluation is guided by their similarity to the properties of interest. This filtering is particularly important for the early identification of scaffolds with favorable ADMET and drug-like properties.

Author
Darren Fayne et al. Senior Research Fellow, Molecular Design Group, School of Biochemistry and Immunology, Trinity College Dublin, Ireland
Lipinski Rule of Five¶
Since published in 1997 the Lipinski rule of five has been an important filter in drug discovery programmes. Compounds will have poor oral absorption if more than two of the following criteria are satisfied.

Author
Darren Fayne et al. Senior Research Fellow, Molecular Design Group, School of Biochemistry and Immunology, Trinity College Dublin, Ireland
Library Design¶
The design and synthesis process for the creation of small molecule libraries requires appropriate planning and execution of multiple and diverse activities as shown below. The conception and development of a library design requires the development of feasible high throughput chemistry, which depends upon the availability of necessary building blocks. After synthesis, compounds must be analyzed, purified, registered and distributed to the appropriate assays and inventories for future research in following up active structures created by this process.
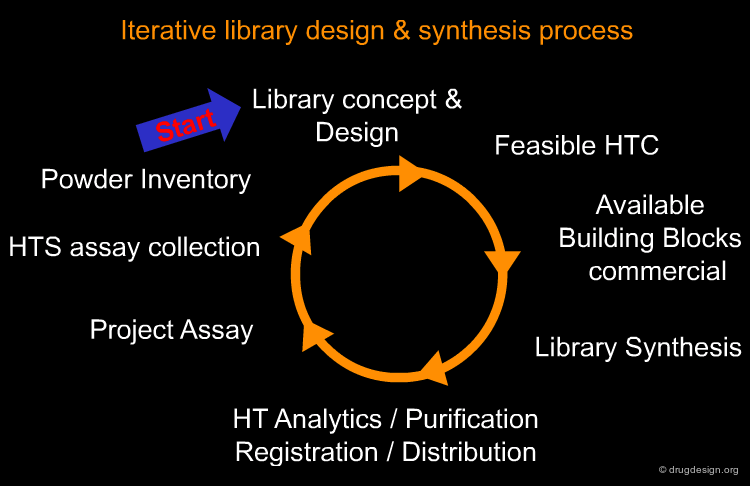
Author
Robert Goodnow Senior Research Leader, Discovery Chemistry, Lead Generation Hoffmann-La Roche Inc., Nutley, New Jersey, USA
Strategies for Library Design Depend Upon Intent¶
The themes which guide library design strategies vary depending upon the type of library one wishes to create. The creation of larger numbers of compounds with a common chemistry method is often more appropriate for building a large HTS collection. However, when targeting hit-to-lead libraries as a follow-up to primary hits detected from an HTS campaign, libraries of fewer numbers are often more useful.
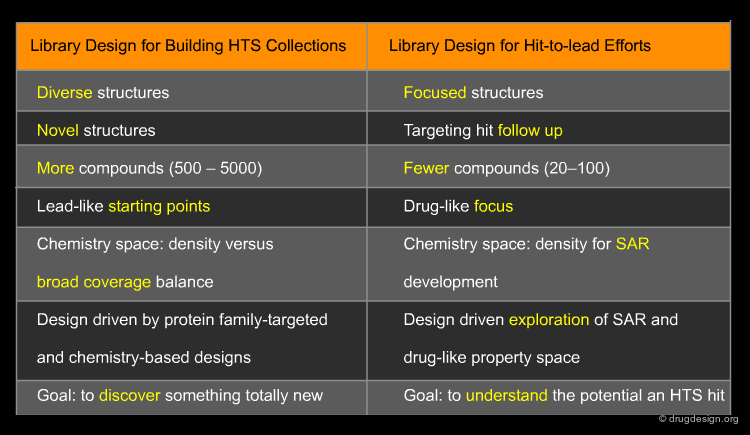
Author
Robert Goodnow Senior Research Leader, Discovery Chemistry, Lead Generation Hoffmann-La Roche Inc., Nutley, New Jersey, USA
HTS Collection¶
Many drug discovery organizations acquire compounds for HTS according to putatively active structural motifs targeting specific protein families such as kinases and GPCRs. The diversity of such protein family targeted subsets is enhanced with compounds resulting from historical medicinal chemistry research, compound purchases and chemistry-based diversity libraries; for such compounds, there is no particular design intent.
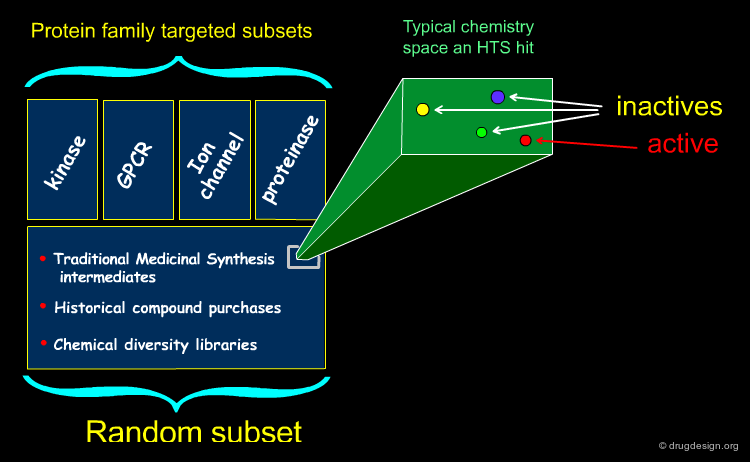
Author
Robert Goodnow Senior Research Leader, Discovery Chemistry, Lead Generation Hoffmann-La Roche Inc., Nutley, New Jersey, USA
Hit-To-Lead Chemistry Space¶
Good, drug-like properties are a constraint on the acquisition of all compounds. When hits are detected, there are often only a few examples. Hit-to-lead library design is one way to increase the density of chemistry space coverage around a particular primary hit.
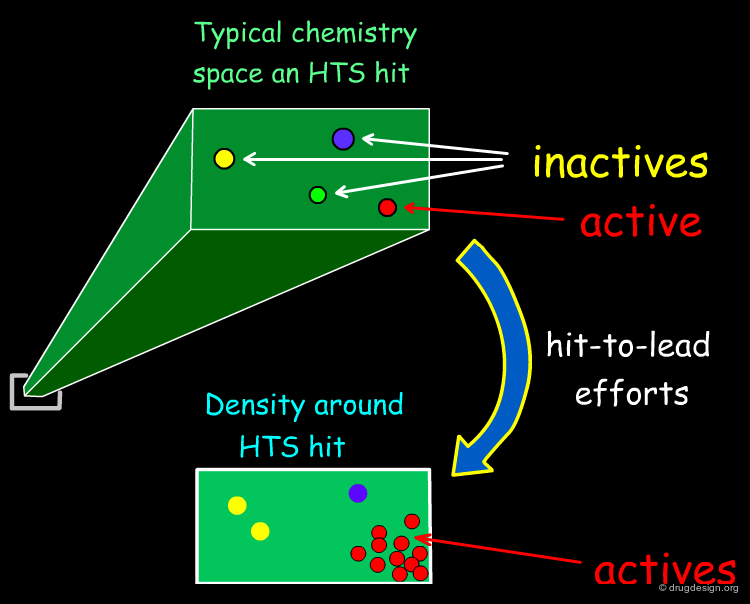
Author
Robert Goodnow Senior Research Leader, Discovery Chemistry, Lead Generation Hoffmann-La Roche Inc., Nutley, New Jersey, USA
Visualizing the Chemistal Space of a Library¶
Chemistry space can be visualized using molecular descriptors. For example BCUT is an abstract molecular descriptor often employed for diversity selection. It combines atomic properties such as charges, polarizabilities, H-bonding capabilities with 2D connectivity and distances.

Author
Robert Goodnow Senior Research Leader, Discovery Chemistry, Lead Generation Hoffmann-La Roche Inc., Nutley, New Jersey, USA
book
Pearlman, RS; Smith, KM Perspectives in Drug Discovery and Design Volumes 9-11 3D QSAR in Drug Design: Ligand/Protein Interactions and Molecular Similarity Springer 1998
Multiple Libraries Diversify a Chemical Space¶
A multi-dimensional BCUT plots is shown for an HTS compound collection of ~200,000 diverse molecules (dark blue dots). Represented with colored dots are collections of virtually created structures according to the same BCUT property analysis. This figure highlights the more extensive chemistry space coverage by many diverse, small molecule libraries as opposed to only a few library designs having many thousands of members.
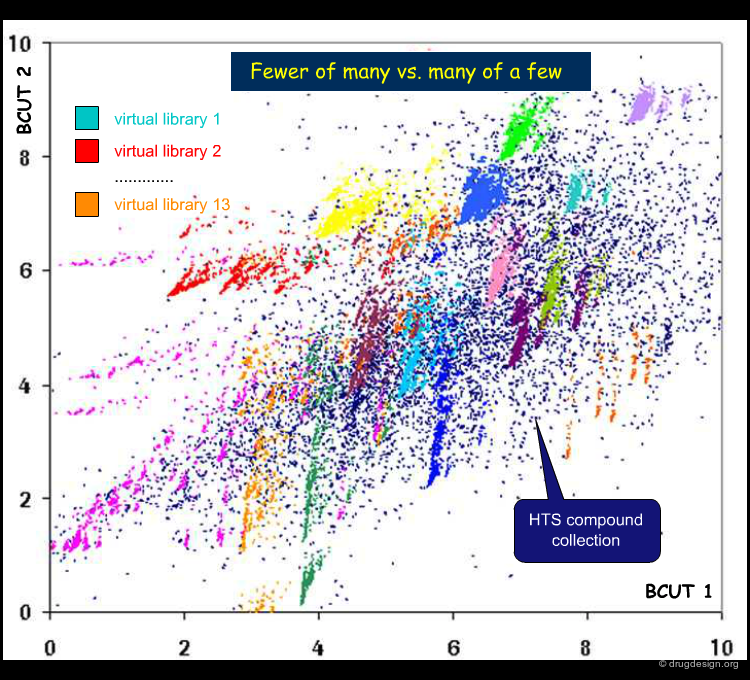
Author
Robert Goodnow Senior Research Leader, Discovery Chemistry, Lead Generation Hoffmann-La Roche Inc., Nutley, New Jersey, USA
book
Pearlman, RS; Smith, KM Perspectives in Drug Discovery and Design Volumes 9-11 3D QSAR in Drug Design: Ligand/Protein Interactions and Molecular Similarity Springer 1998
The Ideal vs Reality of HTC Libraries¶
Although the idea of design, synthesis and assay of small molecule libraries is appealing to many, the reality of successful completion is often quite challenging. Shown below is a correspondence between some of the desired goals for such libraries and frequently encountered limitations. With proper planning to address these limitations, greater success in research using small molecule libraries is likely.

Author
Robert Goodnow Senior Research Leader, Discovery Chemistry, Lead Generation Hoffmann-La Roche Inc., Nutley, New Jersey, USA
Synthesis and Assay of a Kinase Inhibitor Library¶
A library of kinase inhibitors was designed based on a co-crystal structure of a small molecule with a protein kinase. The results of synthesis and assay of small molecule library targeted for kinase inhibition are shown below. A multiple-step, solid-phase synthesis was developed and run to create 1161 structures; the 681 successfully synthesized molecules were assayed for inhibition of human KDR. Against this target, 4 hits having IC50 less than 1µM were found (in yellow in the figure).

Author
Robert Goodnow Senior Research Leader, Discovery Chemistry, Lead Generation Hoffmann-La Roche Inc., Nutley, New Jersey, USA
Biasing for Good Pharmacological and ADME Space¶
With a nearly infinite number of compounds that are possible with modern synthetic methods applied to drug discovery problems, it is important to have a rational process for the target-biased selection of specific templates as well as library design with a given set of diversity reagents (i.e., R groups). Simultaneously, it is important to targeted library design to optimize compounds for good absorption, usually oral. A flow chart below shows the integration of calculated properties for optimizing the balance of pharmacological potency and good ADME property chemistry space.

Author
Robert Goodnow Senior Research Leader, Discovery Chemistry, Lead Generation Hoffmann-La Roche Inc., Nutley, New Jersey, USA
Importance of the Property Analysis¶
The following example illustrates the importance in the analysis of the properties of the virtual molecules. Below is shown a 2-diversity input array of 2400 virtually generated structures designed as inhibitors of a specific enzyme. To study the impact of three selected R2 reagents, the distribution of calculated property scores for each sub-library was analyzed (see histogram).

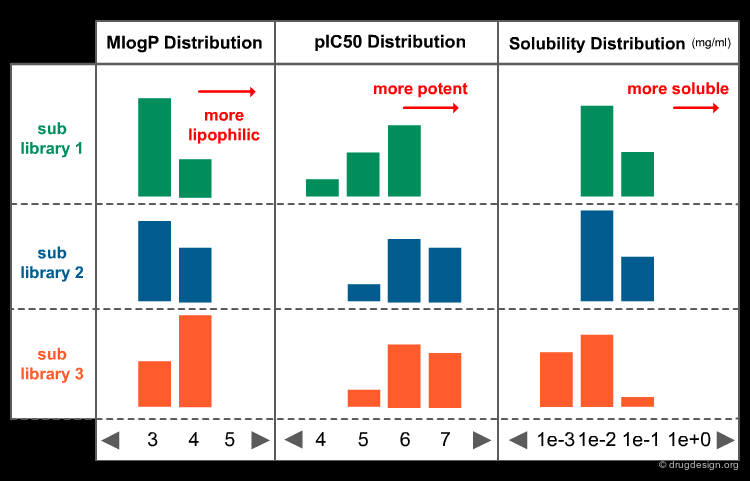
Author
Robert Goodnow Senior Research Leader, Discovery Chemistry, Lead Generation Hoffmann-La Roche Inc., Nutley, New Jersey, USA
Result of the Analyses¶
Although the calculated lipophilicity scores are roughly equivalent, the 1st reagent is predicted to have a lower overall pIC50 (lower target affinity). Additionally, the 3rd diversity reagent results in lower predicted solubility. Thus integrating this information, the 2nd diversity reagent is a better choice than the 1st and 3rd reagents.

Author
Robert Goodnow Senior Research Leader, Discovery Chemistry, Lead Generation Hoffmann-La Roche Inc., Nutley, New Jersey, USA
Library Optimization: R Group Selection Examples¶
Shown below left are examples of substitution patterns which result in compounds of lesser calculated lipophilicities and greater calculated solubilities. By contrast, compounds which contain substitution patterns at right often contribute to the creation of compounds having higher molecular weights, greater lipophiliticities and lower calculated solubilities.

Author
Robert Goodnow Senior Research Leader, Discovery Chemistry, Lead Generation Hoffmann-La Roche Inc., Nutley, New Jersey, USA
Visualization of Library Performance - 1¶
Shown below is the number of primary hits resulting from the high throughput screening of 72 different library designs of various sizes assayed against between 1 and 115 different targets. From this plot, one can state that the HTS of compound libraries of greater numbers results in a greater number of hits over time depending on the protein target of the screen.
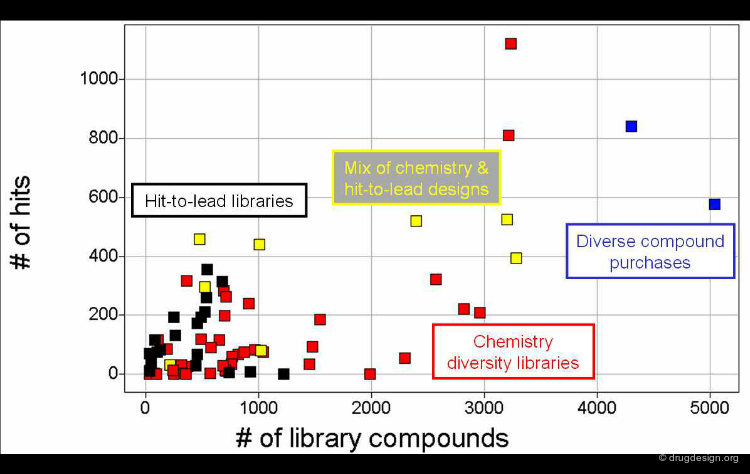
Author
Robert Goodnow Senior Research Leader, Discovery Chemistry, Lead Generation Hoffmann-La Roche Inc., Nutley, New Jersey, USA
Visualization of Library Performance - 2¶
Shown below is the log of the "hit rate" for library designs; the hit rate is defined by the equation below. In this analysis, one can state that the hit rate is enhanced for libraries which are designed around an active starting point, as compared to diverse library designs. The plot is also derived from data comprised of the number of primary hits resulting from the high throughput screening of 72 different library designs of various sizes assayed against between 1 and 115 different targets.

Author
Robert Goodnow Senior Research Leader, Discovery Chemistry, Lead Generation Hoffmann-La Roche Inc., Nutley, New Jersey, USA
ADME/Tox Prediction¶
Half of the failures for drug development were attributed to poor ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) properties: pharmacokinetics (39%) and animal toxicity (11%).

Author
Tingjun Hou Research scientist in the Department of Chemistry and Biochemistry, The University of California at San Diego, USA
articles
Managing the Drug Discovery/Development Interface Kennedy T Drug Discov. Today 2 (10) 1997
Risks in New Drug Development: Approval Success Rates for Investigational Drugs Dimasi JA Clin. Pharmacol. Ther. 69 2001
ADMET in silico modelling: towards prediction paradise? Han van de Waterbeemd and Eric Gifford Nature Reviews Drug Discovery 2 (March) 2003
Rule of Five: Pioneering Work of ADMET Predictions¶
The rule was proposed by Lipinski based on the analysis of 2245 drugs. Rule of five can be considered as a predictor for intestinal absorption. Poor absorption and permeation are more likely to occur when any two of the following rules are satisfied:

Author
Tingjun Hou Research scientist in the Department of Chemistry and Biochemistry, The University of California at San Diego, USA
articles
Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings C. A. Lipinski, F. Lombardo, B. W. Dominy, P. J. Feeney Adv. Drug Del. Rev. 46 2001
Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings Christopher A. Lipinski, Franco Lombardo, Beryl W. Dominy, Paul J. Feeney Adv. Drug Delivery Rev 23 1997
Important ADMET Processes for Theoretical Predictions¶
The ADMET properties colored in red can be predicted in relatively good prediction accuracy.

Author
Tingjun Hou Research scientist in the Department of Chemistry and Biochemistry, The University of California at San Diego, USA
articles
Predictive, computational models of ADME properties Carlson, T. J. and Segall, M. D. Curr. Drug Disc. March 2002
Methods for ADMET Predictions¶
The predictions of the AMDET properties are involved in two aspects of modeling methods: data modeling and molecular modeling. Molecular modeling techniques are used to explore the potential interactions between the small molecules under consideration and proteins known to be involved in ADMET processes, such as cytochrome P450s. Data modeling techniques, especially quantitative structure-activity/property relationship (QSAR/QSPR) approaches, are typically used to construct prediction models based on appropriate descriptors and statistical approaches.

Author
Tingjun Hou Research scientist in the Department of Chemistry and Biochemistry, The University of California at San Diego, USA
The Processes to Develop a ADMET Prediction Model¶
Three basic factors for developing a ADMET prediction model: data, descriptors, and statistical approach for training. Development of a ADMET prediction model involves the following steps: 1. Preparation of the data 2. Optimization of the 3-D structures 3. Calculations of molecular descriptors 4. Training the prediction model based on the training set 5. Validating the prediction capability for the test set.

Author
Tingjun Hou Research scientist in the Department of Chemistry and Biochemistry, The University of California at San Diego, USA
Statistical Approaches used for ADMET Predictions¶
The statistical approach are applied to develop the regression and classification models.

Author
Tingjun Hou Research scientist in the Department of Chemistry and Biochemistry, The University of California at San Diego, USA
Descriptors used for ADMET Predictions¶
Molecular descriptors can be roughly divided into three categories: 1-D (one-dimension), 2-D, and 3-D descriptors. 1-D descriptors are only dependent on the formula of a molecule; 2-D descriptors are obtained from the connectivity or graph of a molecule; 3-D descriptors contain the 3-D geometric information of a molecule. Descriptor set usually includes a group of 2-D and 3-D molecular descriptors.
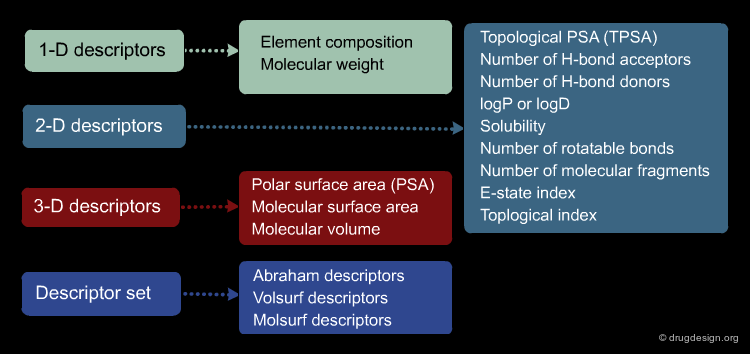
Author
Tingjun Hou Research scientist in the Department of Chemistry and Biochemistry, The University of California at San Diego, USA
Polar Surface Area (PSA): an Important Descriptor¶
Polar Surface Area (PSA) is defined as the surface area associated with the hydrogen-bonding acceptor atoms nitrogen and oxygen and the hydrogen atoms bound to these heteroatoms. Sometimes, sulfur atoms and hydrogen atoms attached to sulfur may also be included. PSA has been widely applied in the predictions of permeability properties, such as Caco-2 permeability, intestinal absorption and blood-brain partitioning.

Author
Tingjun Hou Research scientist in the Department of Chemistry and Biochemistry, The University of California at San Diego, USA
Relationship between PSA and Intestinal Absorption¶
PSA shows an excellent sigmoidal relationship with fractional absorption according to Palm's observations based on a small data set (the left figure). Drugs that are completely absorbed (FA greater than 90%) had a PSA = 60Å2 while drugs that are less than 10% absorbed had a PSA =140 Å2. According to the Hou's observations, TPSA (topological polar surface area) does not have an excellent relationship with fractional absorption based on a large data set of 455 molecules (the right figure).

Author
Tingjun Hou Research scientist in the Department of Chemistry and Biochemistry, The University of California at San Diego, USA
articles
ADME Evaluation in Drug Discovery. 7. Prediction of Oral Absorption by Correlation and Classification Tingjun Hou, Junmei Wang, Wei Zhang, and Xiaojie Xu Journal of Chemical Information and Modeling 47 2007
Polar Molecular Surface Properties Predict the Intestinal Absorption of Drugs in Humans Katrin Palm, Patric Stenberg, Kristina Luthman, Per Artursson Pharmaceutical Research 14 (5) 1997
logD: Another Important Descriptor¶
The apparent coefficient, logD, at pH=6.5 shows obvious correlation with intestinal absorption. According to the figure, logD6.5 = -3.2 may be identified as a rough bound to identify the compounds with a FA smaller than 10% from the others.
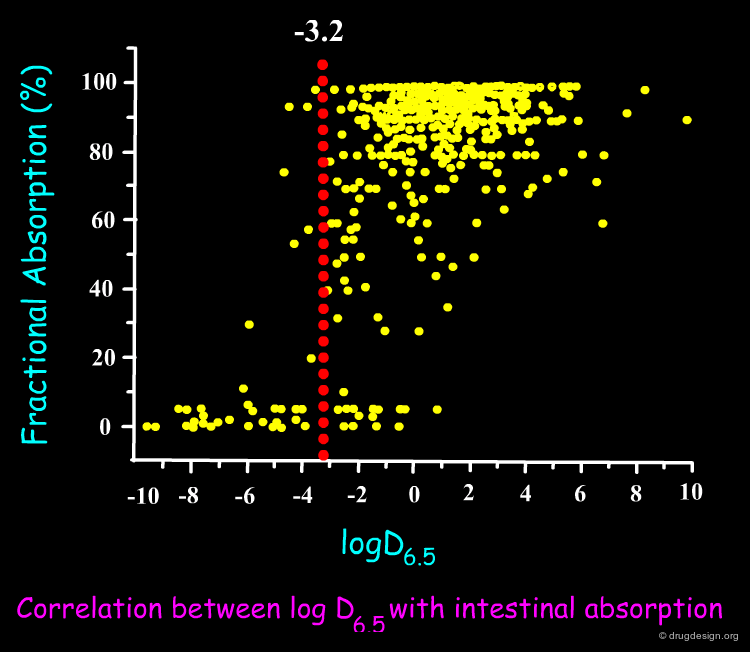
Author
Tingjun Hou Research scientist in the Department of Chemistry and Biochemistry, The University of California at San Diego, USA
articles
ADME evaluation in drug discovery. 8. The prediction of human intestinal absorption by a support vector machine Hou T, Wang J and Li Y. J Chem Inf Model 47 (6) 2007
Prediction Models for the ADMET Properties¶
The prediction models for metabolism are usually developed based on molecular modeling, while most of the prediction models for the other ADMET properties are developed based on data modeling.

Author
Tingjun Hou Research scientist in the Department of Chemistry and Biochemistry, The University of California at San Diego, USA
Regression Models for ADMET Predictions¶
Several representative regression models for predicting Caco-2 permeability, intestinal absorption (%FA), and blood-brain partitioning (logBB) are shown below.

Author
Tingjun Hou Research scientist in the Department of Chemistry and Biochemistry, The University of California at San Diego, USA
articles
ADME Evaluation in Drug Discovery. 5. Correlation of Caco-2 Permeation with Simple Molecular Properties T. J. Hou, W. Zhang, K. Xia, X. B. Qiao, and X. J. Xu J Chem Inf Comput Sci 44 (5) 2004
ADME Evaluation in Drug Discovery. 3. Modeling Blood-Brain Barrier Partitioning Using Simple Molecular Descriptors T. J. Hou and X. J. Xu J Chem Inf Comput Sci 43 (6) 2003
ADME evaluation in drug discovery. 8. The prediction of human intestinal absorption by a support vector machine Hou T, Wang J and Li Y. J Chem Inf Model 47 (6) 2007
Classification Models for ADMET Predictions¶
A classification model to classify 481 compounds into poor and good intestinal absorption classes based on recursive partitioning (RP) is shown below. The model can correctly identify 95.9% (71/74) of the compounds in class 1 and 96.1% (391/407) of the compounds in class 2.

Author
Tingjun Hou Research scientist in the Department of Chemistry and Biochemistry, The University of California at San Diego, USA
articles
ADME evaluation in drug discovery. 8. The prediction of human intestinal absorption by a support vector machine Hou T, Wang J and Li Y. J Chem Inf Model 47 (6) 2007
Structure Models for ADMET Predictions¶
A substrate pharmacophore model developed by Chang et al. for the efflux transporter protein P-gp is shown below (using the Catalyst software). Two compounds: Gleevec (top) and Curcumin (bottom), are mapped to common hydrophobic (blue) and H-bond acceptor (green) features.
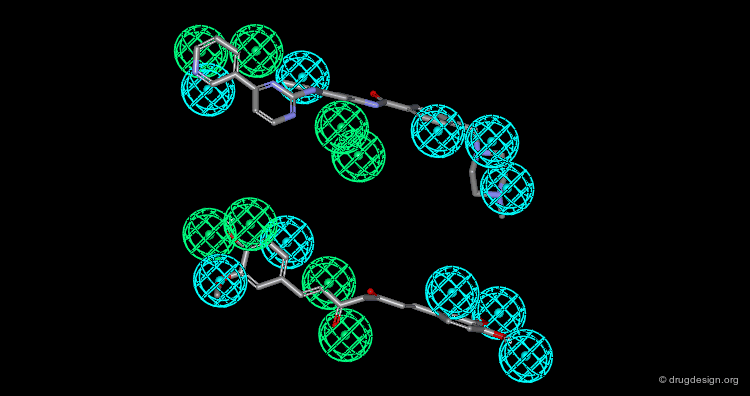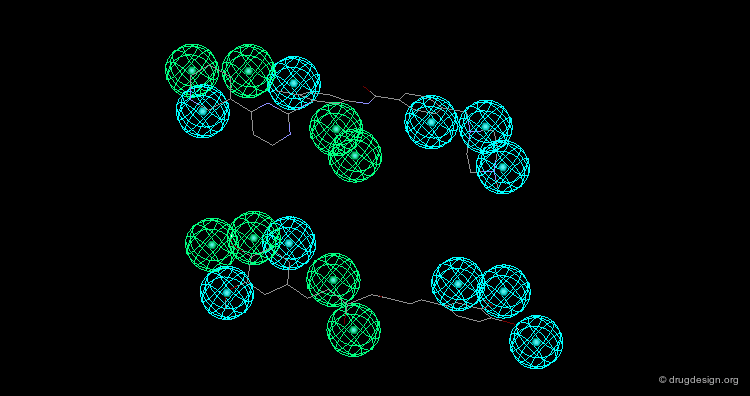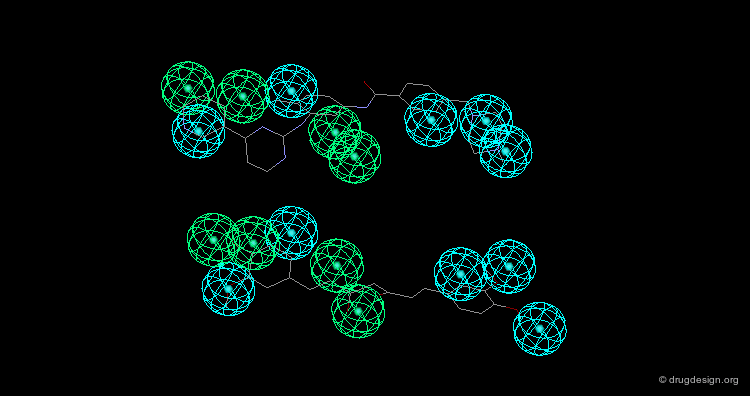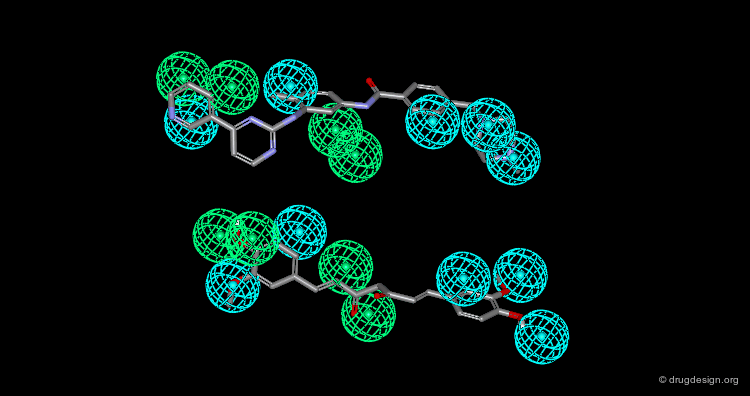

Author
Tingjun Hou Research scientist in the Department of Chemistry and Biochemistry, The University of California at San Diego, USA
articles
Pharmacophore-based discovery of ligands for drug transporters Cheng Chang, Sean Ekins, Praveen Bahadduri and Peter W. Swaan Advanced Drug Delivery Reviews 2006 58 (12-13)
Computer-Assisted Structure Elucidation¶
This section deals with "small molecule" structure elucidation, a part of the process for the discovery of new bio-active molecules. For example, natural product chemists continuously isolate compounds that are engaged in activity screening tests. The structure determination of a biologically active compound is a mandatory step in its potent development as a lead. For the same purpose, the structure of synthetic substances must also be firmly assessed. Computers play an important role at various stages of structure elucidation.
Author
Jean Marc Nuzillard Directeur de recherches au CNRS, Institut de Chimie Moleculaire de Reims, France
Structure and Isomers¶
A single elemental composition formula such as C2H60 accounts for both dimethyl ether and ethanol, two substances that present different chemical functions and therefore different physical properties. They are constitution isomers. Glucose is present in water as α and β anomeric forms. Their chemical functions are the same, they have the same planar (2D) structure but they may react differently and present different physical properties as they have different 3D shapes. The α and β forms are stereo-isomers. At the finest detail level, one may want to determine conformational state and/or stereo-isomer populations.

Author
Jean Marc Nuzillard Directeur de recherches au CNRS, Institut de Chimie Moleculaire de Reims, France
Physical Characterization of Organic Molecules¶
X-ray crystallography apart, the physical techniques that are used for the characterization of organic molecules do not directly provide the "shape" of the molecule but structural clues that must be considered together by the chemist to build a likely structure. Infra-Red (IR), Ultra-Violet (UV) and Nuclear Magnetic Resonance (NMR) spectroscopic techniques exploit the vibrational, electronic and nuclear transitions between energy levels within molecules. The molecular structural features are thus revealed by the changes functional groups cause to energy transitions.

Author
Jean Marc Nuzillard Directeur de recherches au CNRS, Institut de Chimie Moleculaire de Reims, France
Mass Spectrometry (MS)¶
In a mass spectrometer the molecules are transferred to a gas phase and ionized. The ions and their eventual fragments are analyzed according their mass to charge (m/z) ratio. The high resolution mass spectrum of a compound directly gives access to its elemental composition, an information of high importance for structure elucidation, either computer-assisted or carried out "by hand". Fragment ions analysis also provides structural information on the studied molecule. MS fragmentation is a key process for the analysis of biopolymers, but is often of limited interest in computer-assisted structure elucidation.

Author
Jean Marc Nuzillard Directeur de recherches au CNRS, Institut de Chimie Moleculaire de Reims, France
X-ray Crystallography¶
Structural X-ray crystallography applies to compounds for which monocrystals can be obtained and is mainly used when other techniques have failed to provide a satisfactory solution. X-ray crytallography is a highly computerized technique, as electron density map construction requires 3D Fourier transformation computations and the resolution of the mathematically difficult "phase problem".

Author
Jean Marc Nuzillard Directeur de recherches au CNRS, Institut de Chimie Moleculaire de Reims, France
The Special Role of NMR Spectroscopy¶
NMR spectroscopy individually characterizes atom nuclei according to the density repartition of surrounding electrons. Each NMR-active nucleus (1H, 13C, 15N, ...) inside a molecule is thus described by a numerical value known as its chemical shift. Through-bond magnetic interactions between nuclei are measured by a coupling constant value. It is significantly different from zero when the coupled nuclei are separated by a small number of chemical bonds. This provide an unvaluable 2D structural information when combined with chemical shift interpretation. Experimentally, all scalar couplings of a molecular are revealed by two-dimensional (2D) NMR techniques.

Author
Jean Marc Nuzillard Directeur de recherches au CNRS, Institut de Chimie Moleculaire de Reims, France
Spectroscopy and Computers¶
Computers are nowadays in all spectrometers. At low-level, they are used to drive the hardware and to acquire raw data in numerical format. IR-FT and NMR spectrometers also include dedicated software for spectra calculation by Fourier transformation (FT). Spectrum interpretation is a high level task that is presently performed by human intelligence, knowledge and experience. Attempts to formalize and reproduce the way chemists do it is as old as computer artificial intelligence. Computer-Assisted Structure Elucidation (CASE) systems have evolved to a point they can solve non-trivial real-world problems.
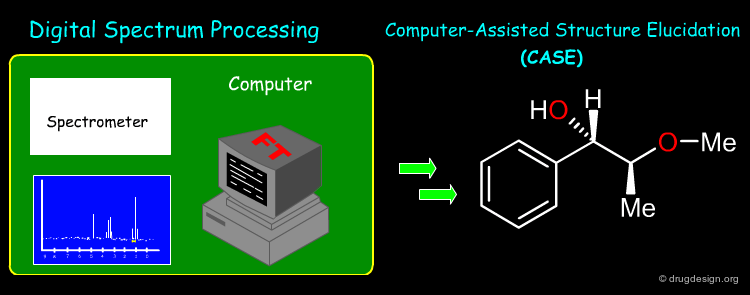
Author
Jean Marc Nuzillard Directeur de recherches au CNRS, Institut de Chimie Moleculaire de Reims, France
Dereplication¶
Dereplication is the process that avoids one to solve a structure search problem that has already been solved by others. It is carried out by comparison of the physico-chemical properties of a substance with those stored in a structure/properties pair library. Properties may simply consist of a chromatographic retention time and a mass spectrum, as provided by an hyphenated HPLC-MS purification system. Computers are of critical importance for the efficient storage and retrieval of diverse property measurements. Unmatched compounds must then be submitted to a thorough analytical procedure in view of their structure elucidation.

Author
Jean Marc Nuzillard Directeur de recherches au CNRS, Institut de Chimie Moleculaire de Reims, France
Functions of a Typical CASE System¶
The functions of a typical CASE system are: (1) data reduction, (2) structure generation, (3) solution validation/ranking. Not all spectral point values with an IR or a NMR spectrum are of interest. The extraction of pertinent spectral features (peak location and intensity) is named data reduction. Structure generation is achieved by selecting molecular fragments that match with the reduced data and by assembling them into molecular structures. In the frequent situation the problem has more than one solution, the candidate structures should be sorted in decreasing likelihood order.

Author
Jean Marc Nuzillard Directeur de recherches au CNRS, Institut de Chimie Moleculaire de Reims, France
Advantages and Drawbacks¶
The most important advantage brought by CASE systems is their absence of preconception about the kind of result that has to be found. A lot of time may be wasted if one persists to search for a structural feature that is not present in the molecule under investigation. The drawback in using CASE systems is they lack the year-long experience of human beings that makes particular problems trivial to solve. Users must be trained to efficiently use CASE systems. Depending on feature abundance and ergonomy, the learning curve can be steep enough to refrain chemists to use them.

Author
Jean Marc Nuzillard Directeur de recherches au CNRS, Institut de Chimie Moleculaire de Reims, France
Constraint Generation from Spectral Data¶
A correct elemental formula is necessary to start solving a structure problem, even though it is not always available in real world situations. The complexity of molecules that can be treated on the basis of 1H NMR only (1D and 2D) is rather limited and requires complementary information from 13C and eventually 15N NMR data. The 2D HSQC and HMBC NMR spectra have tremendously changed the way chemist solve small molecule structure problems. Accordingly, the power of CASE system has considerably increased with the handling of structure constraints that lie in these heteronuclear chemical shift correlation spectra.
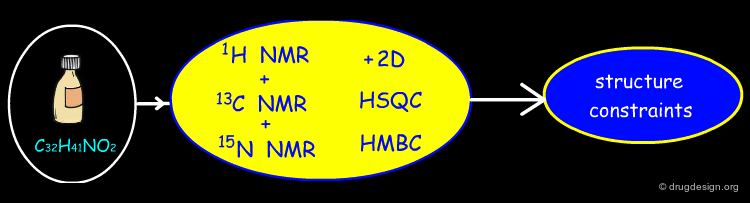
Author
Jean Marc Nuzillard Directeur de recherches au CNRS, Institut de Chimie Moleculaire de Reims, France
NMR Chemical Shift, UV and IR Constraints¶
NMR chemical shifts provide atom-centered molecular fragments, in a way that is far from being univocal. This means that the presence of some fragment may be ruled out as judged incompatible with a given chemical shift value. A high number of such constraints may however be sufficient to propose a reasonably low number of solutions, if not only one when the structure is not too complex. UV and IR spectra provide lists of fragments that are compatible with the observed light absorption frequencies and are complementary to NMR chemical shifts.

Author
Jean Marc Nuzillard Directeur de recherches au CNRS, Institut de Chimie Moleculaire de Reims, France
NMR Couplings¶
Scalar NMR couplings concern atom pairs whose members are separated by a limited number of chemical bonds. The existence of a non negligible scalar coupling interaction between two atoms reveals a structure fragment in which they are identified by their chemical shift. Such couplings concern pairs of atoms, either of the same element (homonuclear) or of different elements (heteronuclear). A scalar coupling through n bonds is referred to as a nJ coupling. Dipolar couplings are direct, through-space interactions between atom nuclei that are close to each other. Their provide most of the available information about the 3D structures of molecules. All types of couplings are efficiently detected by multi-dimensional NMR spectra.

Author
Jean Marc Nuzillard Directeur de recherches au CNRS, Institut de Chimie Moleculaire de Reims, France
Constraints from 2D NMR --> Flat Structures¶
The search for the "flat" structure (i.e. with no stereochemical information) of a small organic molecule is basically achieved by inspection of three 2D NMR experiments. The COSY, HSQC and HMBC spectra respectively reveal the 1H homonuclear couplings, the 1J 1H-X and the nJ (n > 1) heteronuclear couplings. X stands most often for 13C as information on carbon atoms is of the highest importance for organic molecules. However, 15N NMR spectroscopy is also pertinent for many biologically active compounds.
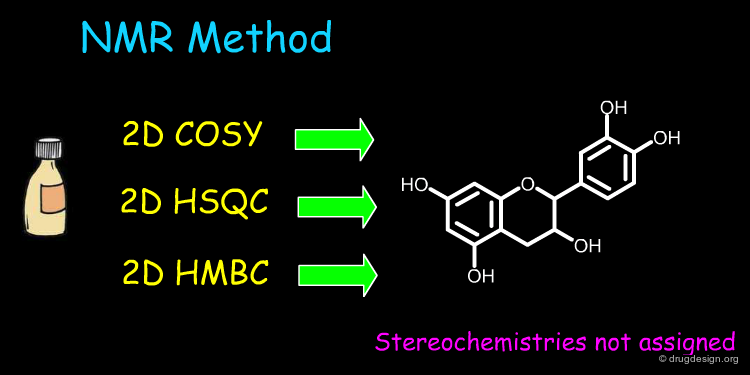
Author
Jean Marc Nuzillard Directeur de recherches au CNRS, Institut de Chimie Moleculaire de Reims, France
Constraints from 2D NMR --> Stereochemistry¶
The through-bond proximity relationships of a molecule represent an important part of the constraint set used by NMR-based CASE systems. The 2D NOESY and ROESY 1H NMR spectra bring through-space proximity relationships that are needed to propose likely structures with their stereochemistry.

Author
Jean Marc Nuzillard Directeur de recherches au CNRS, Institut de Chimie Moleculaire de Reims, France
Generating Solutions with Structure Generators¶
Structure generators are molecular fragment assemblers. They take into account the basic rules of organic chemistry and the user-supplied constraints that derive from the spectral data of an unknown molecule. Structure generators are expected to provide a complete set of solutions for a given problem, possibly without redundancies. As for any software, the quality of the output reflects the quality of the input, namely the data that is available. Inconsistent data, such as wrong user misconception on what the result should be, leads to solution search failure.

Author
Jean Marc Nuzillard Directeur de recherches au CNRS, Institut de Chimie Moleculaire de Reims, France
General Considerations on Structure Generators¶
Structure generation may be either combinatorial or stochastic. The former strategy is the most common and relies on the systematic assembly of molecular fragments that derive from spectral data. A single spectroscopic information may be compatible with many fragments. The generator has thus to manage at best the order in which individual data pieces are used, so that no combinatorial happens. Alternatively, stochastic generators are inspired from optimization techniques by simulated annealing. A set of randomly drawn initial structures is improved by atom permutations until the structural constraints are satisfied at best.

Author
Jean Marc Nuzillard Directeur de recherches au CNRS, Institut de Chimie Moleculaire de Reims, France
Validating the Results¶
When a structure generator produces more than one solution that is compatible with the provided structural constraints, the solutions must be ranked in the order of decreasing likelihood. Solution evaluation is achieved by measuring the "distance" between experimental molecular properties and predicted ones. The accurate computation of molecular properties (including spectroscopic properties) from structure can only be performed on complete structures and not on the intermediate fragments that are formed during the resolution process. For this reason, spectrum prediction tools are integrated in CASE systems.

Author
Jean Marc Nuzillard Directeur de recherches au CNRS, Institut de Chimie Moleculaire de Reims, France
NMR Spectrum Prediction¶
The structure generators produce connectivity graphs between atoms that correspond to 2D molecular structures. Even though molecules are 3D objects, NMR chemical shift prediction can be carried out from 2D structures, with some accurary limitations. All methods rely on the considerable amount of data that has been recorded over the last decades. Atom descriptors within known molecules, such as HOSE codes, are keys to the possible chemical shift values of atoms in unknown molecules. Other methods, such as increment-based calculations or prediction by artificial neural networks rely on the exploitation of experimental data collections.

Author
Jean Marc Nuzillard Directeur de recherches au CNRS, Institut de Chimie Moleculaire de Reims, France
Advanced NMR Spectrum Prediction¶
NMR spectrum prediction from a 3D structure requires the latter to be known. NOESY or ROESY data are used to constrain the generation of possible 3D structures from a 2D one, along with geometry-dependent scalar coupling data. Again, the ranking of 3D structures is achieved by comparison of experimental and calculated spectral parameters. NMR chemical shift data banks that handle the 3D nature of molecules exist and are used for this purpose.

Author
Jean Marc Nuzillard Directeur de recherches au CNRS, Institut de Chimie Moleculaire de Reims, France
Ab-Initio Calculations of Shifts and Coupling¶
Another approach relies on computational chemistry techniques to calculate chemical shifts and coupling constants by ab initio methods. Computation times are longer than for data bank-based methods by many order of magnitudes. However, ab initio methods should become more commonly used in a near future.

Author
Jean Marc Nuzillard Directeur de recherches au CNRS, Institut de Chimie Moleculaire de Reims, France
De-Novo Design¶
Drug design approaches are extensively presented in this course. Two major methods are used in de-novo design: the Structure-Based and the Ligand-Based approaches. In this section we briefly present and illustrate these methods.

Structure-Based Design¶
When the 3D structure of the target protein associated to the disease concerned is available (experimentally or generated by homology modeling) it is possible to use a structure-based approach. In the following pages we present three possible strategies.

Docking using Libraries of Compounds¶
With the advent of efficient and simple docking methods it has become possible to screen entire libraries of molecules, each compound being defined as a set of low energy conformers. Below is shown a BACE-1 inhibitor derived from a hit discovered by this method, known as high-throughput screening.

articles
Structure-based design of potent and selective cell-permeable inhibitors of human beta-secretase (BACE-1) Stachel SJ, Coburn CA, Steele TG, Jones KG, Loutzenhiser EF, Gregro AR, Rajapakse HA, Lai MT, Crouthamel MC, Xu M, Tugusheva K, Lineberger JE, Pietrak BL, Espeseth AS, Shi XP, Chen-Dodson E, Holloway MK, Munshi S, Simon AJ, Kuo L, Vacca JP. J Med Chem. 47 2004
Automated Construction Methods¶
The purpose of construction programs is to discover a new chemical framework that fits to the active site of the target receptor or enzyme. Some methods are based on an existing moiety and additional fragments are appended by a step-by-step build up procedure. Other methods consist of assembling novel molecules from pieces that are positioned optimally in favorable regions of the active site. The difficulty of the approach remains on the synthetic feasibility of the molecules constructed.

articles
The Computer Program LUDI: a New Method for the De-Novo Design of Enzyme Inhibitor Leads Bohm H-J J. Comput. Aided Mol. Des. 6 1992
LUDI: Rule-Based Automatic Design of New Substituents for Enzyme Inhibitor Leads Bohm H-J J. Comput. Aided Mol. Des. 6 1992
Automated Site-Directed Drug Design: the Generation of a Basic Set of Fragments to be Used for Automated Structure Assembly Chau PL and Dean PM J. Comput. Aided Mol. Des. 6 1992
PRO-LIGAND: an Approach to De Novo Molecular Design. 1. Application to the Design of Organic Molecules Clark DE, Frenkel D, Levy SA, Li J, Murray CW, Robson B, Waszkowycz B and Westhead DR J. Comput. Aided Mol. Des. 9 1995
De Novo Design of Enzyme Iinhibitors by Monte Carlo Ligand Generation Gehlhaar DK, Moerder KE, Zichi D, Sherman CJ, Ogden RC and Freer ST J. Med. Chem. 38 1995
SPROUT, HIPPO and CAESA: Tools for De Novo Structure Generation and Estimation of Synthetic Accessibility Gillet VJ, Myatt G, Zsoldos Z and Johnson P Persp. Drug Discov. Des. 3 1995
CAVEAT: a Program to Facilitate the Design of Organic Molecules Lauri G and Bartlett PA J. Comput. Aided. Mol. Des. 8 1994
Automatic Creation of Drug Candidate Structures Based on Receptor Structure: Starting Point for Artificial Lead Generation Nishibata Y and Itai A Tetrahedron 47 1991
Confirmation of Usefulness of a Structure Construction Program Based on Three-Dimensional Receptor Structure for Rational Lead Generation Nishibata Y, Itai A J. Med. Chem. 36 1993
CONCERTS: Dynamic Connection of Fragments as an Approach to De Novo Ligand Design Pearlman DA and Murcko MA J. Med .Chem. 39 1996
A Genetic Algorithm for Structure-Based De Novo Design Pegg SC, Haresco JJ, Kuntz ID J. Comput. Aided Mol. Des. 15 2001
BUILDER v.2: Improving the Chemistry of a De-Novo Design Strategy Roe DC and Kuntz ID J. Chem. Inf. Comput. Sci. 9 1995
GroupBuild: a Fragment-Based Method for De-Novo Drug Design Rotstein SH, Murcko MA J. Med. Chem. 36 1993
Manual Design (Structure-Based)¶
Manual design remains a simple approach for a medicinal chemist looking for new ideas. Based on the X-ray structure of a ligand complexed with the target protein considered, it is possible to exploit this knowledge by modeling analogs of novel mimics that exploit optimally good interactions with the protein. The example below illustrates the discovery of a cathepsin K compound designed to obtain improved synthetic accessibility and oral bioavailability.
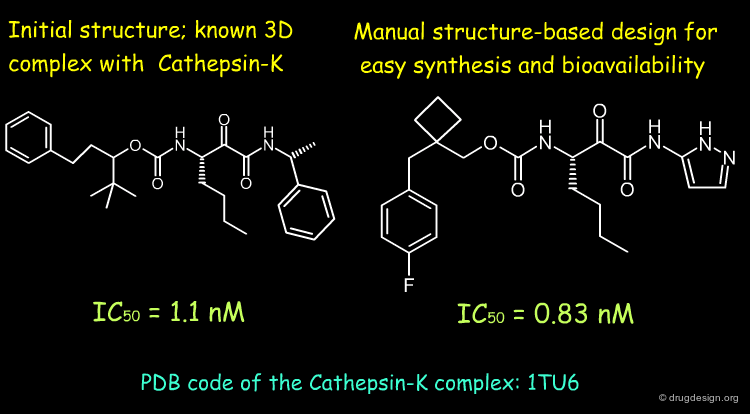
articles
Potent and selective P2-P3 ketoamide inhibitors of cathepsin K with good pharmacokinetic properties via favorable P1', P1, and/or P3 substitutions Barrett DG, Catalano JG, Deaton DN, Hassell AM, Long ST, Miller AB, Miller LR, Shewchuk LM, Wells-Knecht KJ, Willard DH Jr, Wright LL. Bioorg and Medicinal Chemistry Letters 14 2004
Ligand-Based Design¶
When at least one active compound is available but the 3D structure of the target protein is not known, the strategy for designing new compounds is "ligand-based" or "pharmacophore-based" drug design. Three different strategies associated to this approach are presented in the following pages.

Virtual Screening¶
In a ligand-based perspective virtual screening can be made using either a 2D database of compounds or a 3D database.
Virtual Screening with 2D Databases¶
It is very simple to do 2D substructure search using for example a corporate, or a commercial 2D database of compounds such as ZINC. In the example illustrated below, the 2D search aimed at finding bioisosteres of a benzothiazole heterocycle.

articles
ZINC--a free database of commercially available compounds for virtual screening. Irwin JJ, Shoichet BK. J Chem Inf Modeling. 45 2005
Virtual Screening with 3D Databases¶
An example of 3D search is shown here. The 3D pharmacophore is indicated in the view, together with the hits obtained by the search. This project aims at the discovery of novel dopamine transporter inhibitors.
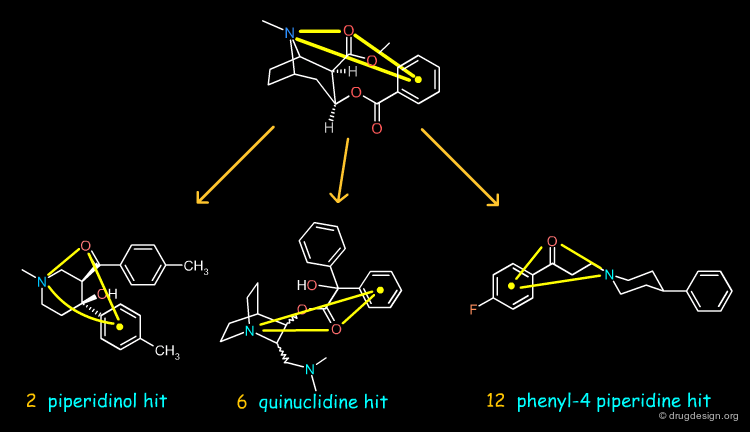
articles
Discovery of a novel dopamine transporter inhibitor, 4-hydroxy-1-methyl-4-(4-methylphenyl)-3-piperidyl 4-methylphenyl ketone, as a potential cocaine antagonist through 3D-database pharmacophore searching. Molecular modeling, structure-activity relationships, and behavioral pharmacological studies. Wang S, Sakamuri S, Enyedy IJ, Kozikowski AP, Deschaux O, Bandyopadhyay BC, Tella SR, Zaman WA, Johnson KM. J Med Chem. 43(3) 2000
Automated Construction Methods¶
The purpose of de novo automated construction approaches is to find appropriate spacers to assemble disconnected elements. The solutions show alternatives for positioning the same key fragments and therefore provide structural diversity. An example of molecule generated by a computer program (NEWLEAD) that mimick a steroid compound is given below.

articles
The NEWLEAD Program : A New Method for the Design of Candidate Structures from Pharmacophoric Hypotheses V. Tschinke and N.C. Cohen J. Med. Chem. 36 1993
Manual Design (Ligand-Based)¶
Manual design remains the more natural way for a medicinal chemist for designing a new compound, based on the 2D and/or 3D structure of known active molecules. The following illustrates the intelligent design of an anthranilamide scaffold conceived to mimic an initial anilinophtalazine compound.

Essential Algorithms in Cheminformatics¶
Need of Algorithms in Cheminformatics¶
Chemical structures share similar patterns. To search, detect and compare molecules in parts or as a whole, we need some tools to perceive and handle these patterns. This is the role of algorithms that are fixed and reusable procedures which consider each situation for a particular problem. It is also time-saving because, with the aid of the computers, it enables to handle numerous and complex cases.

Author
Y Jane Tseng Associate Professor, Laboratory of Computational Molecular Detection and Design, Graduate Institute of Biomedical Electronics and Bioinformatics, Department of Computer Science and Information Engineering, National Taiwan University, Taiwan
Algorithms Presented in this Section¶
Many algorithms are repeatedly used in cheminformatic applications, some of them are described in the present section and are listed in the figure below.

Author
Y Jane Tseng Associate Professor, Laboratory of Computational Molecular Detection and Design, Graduate Institute of Biomedical Electronics and Bioinformatics, Department of Computer Science and Information Engineering, National Taiwan University, Taiwan
Ring Searches¶
Rings are important features of chemical structures. The aim of ring search algorithms is to start with the connectivity matrix of a molecule and to look if there are rings in the structure, and what are those rings. Try to recognize them from the connectivity table in the clavulanic acid example below! The algorithm must find three rings that are contained in the encoded structure.


Author
Y Jane Tseng Associate Professor, Laboratory of Computational Molecular Detection and Design, Graduate Institute of Biomedical Electronics and Bioinformatics, Department of Computer Science and Information Engineering, National Taiwan University, Taiwan
articles
Review of Ring Perception Algorithms for Chemical Graphs Downs, G.; Gillet, V.; Holliday, J.; Lynch, M. J. Chem. Inf. Comput. Sci 29 1989
Efficient Exact Solution of the Ring Perception Problem Renzo Balducci and Robert S. Pearlman Journal of Chemical Information and Computer Science 34 (4) 1994
Rings Present in a Structure¶
A simple formula gives the number of smalest rings in a structure from the number of nodes (atoms) and edges (number of distinct atom-to-atom connections) present in the system. The general formula is given below and illustrated with two examples.

Author
Y Jane Tseng Associate Professor, Laboratory of Computational Molecular Detection and Design, Graduate Institute of Biomedical Electronics and Bioinformatics, Department of Computer Science and Information Engineering, National Taiwan University, Taiwan
articles
Efficient Exact Solution of the Ring Perception Problem Renzo Balducci and Robert S. Pearlman Journal of Chemical Information and Computer Science 34 (4) 1994
Ring Perception. A New Algorithm for Directly Finding the Smallest Set of Smallest Rings from a Connection Table Bo Tao Fan, Annick Panaye, Jean-Pierre Doucet, and Alain Barbu Journal of Chemical Information and Computer Science 33 1993
Ring Perception. A New Algorithm for Directly Finding the Smallest Set of Smallest Rings from a Connection Table Bo Tao Fan, Annick Panaye, Jean-Pierre Doucet, and Alain Barbu Journal of Chemical Information and Computer Science 33 1993
Ring Perception Using Breadth-First Search John Figueras Journal of Chemical Information and Computer Science 36 (5) 1996
A New Algorithm for Exhaustive Ring Perception in a Molecular Graph Th. Hanser, Ph. Jauffret, and G. Kaufmann Journal of Chemical Information and Computer Science 36 1996
Fast algorithm for ring perception Ludek Matyska Journal of Computational Chemistry 9 (5) 1988
Smallest Set of Smallest Rings: Example¶
For many applications it is important to recognize only the smallest rings (also called "fundamental rings"). In the steroid example below, the algorithm is expected to recognize (based on the connectivity matrix) only rings A, B, C and D of the estradiol structure, and not others such as, for example, the 10-membered and 17-membered rings displayed in the figure.

Author
Y Jane Tseng Associate Professor, Laboratory of Computational Molecular Detection and Design, Graduate Institute of Biomedical Electronics and Bioinformatics, Department of Computer Science and Information Engineering, National Taiwan University, Taiwan
articles
Ring Perception. A New Algorithm for Directly Finding the Smallest Set of Smallest Rings from a Connection Table Bo Tao Fan, Annick Panaye, Jean-Pierre Doucet, and Alain Barbu Journal of Chemical Information and Computer Science 33 1993
Ring Perception Using Breadth-First Search John Figueras Journal of Chemical Information and Computer Science 36 (5) 1996
Smallest Set of Smallest Rings: Algorithm¶
The algorithm for finding the smallest rings in a structure is as follows: (1) choose each atom in the structure to be a starting atom; (2) search for the smallest ring by eliminating reducible atoms and finally, (3) the result is the smallest set of smallest rings.

Author
Y Jane Tseng Associate Professor, Laboratory of Computational Molecular Detection and Design, Graduate Institute of Biomedical Electronics and Bioinformatics, Department of Computer Science and Information Engineering, National Taiwan University, Taiwan
articles
Ring Perception. A New Algorithm for Directly Finding the Smallest Set of Smallest Rings from a Connection Table Bo Tao Fan, Annick Panaye, Jean-Pierre Doucet, and Alain Barbu Journal of Chemical Information and Computer Science 33 1993
Ring Perception Using Breadth-First Search John Figueras Journal of Chemical Information and Computer Science 36 (5) 1996
Fast algorithm for ring perception Ludek Matyska Journal of Computational Chemistry 9 (5) 1988
A New Algorithm for Exhaustive Ring Perception in a Molecular Graph Th. Hanser, Ph. Jauffret, and G. Kaufmann Journal of Chemical Information and Computer Science 36 1996
Fast algorithm for ring perception Ludek Matyska Journal of Computational Chemistry 9 (5) 1988
Aromaticity Detection¶
The algorithm for aromaticity ring detection is based on the following rules: (1) the molecule must contain a planar ring system; (2) all atoms of the ring must be hybridized sp2 and (3) the total number of π electrons must be equal to (4n+2), n being even (Huckel rules). Below are shown examples of aromatic and non-aromatic structures.

Author
Y Jane Tseng Associate Professor, Laboratory of Computational Molecular Detection and Design, Graduate Institute of Biomedical Electronics and Bioinformatics, Department of Computer Science and Information Engineering, National Taiwan University, Taiwan
articles
Aromaticity and conjugation Milan Randic Journal of the American Chemical Society 99 (2) 1977
wikipedia
Fingerprinting¶
Usually a fingerprinting algorithm examines the molecule and generates a set of patterns. Below is shown a Daylight fingerprint for the OC=CN molecule, as an example.

Author
Y Jane Tseng Associate Professor, Laboratory of Computational Molecular Detection and Design, Graduate Institute of Biomedical Electronics and Bioinformatics, Department of Computer Science and Information Engineering, National Taiwan University, Taiwan
Hash Codes: a Unique Compressed Code¶
A hash code is a unique number which describes and identifies molecular data structures in chemistry such as atoms and bonds, and it is characteristic of an individual chemical structure. The algorithm sketched below produces a highly compressed code dependent only on the input information (A), such as molecular weight or empirical formula.

Author
Y Jane Tseng Associate Professor, Laboratory of Computational Molecular Detection and Design, Graduate Institute of Biomedical Electronics and Bioinformatics, Department of Computer Science and Information Engineering, National Taiwan University, Taiwan
Hash Codes: the Key Transformation¶
The hash procedure (key transformation) computes a number of storage addresses from alphabetic, numeric, or alphanumeric keys (B). The hash code includes no data information: it is only used as a key to the storage address of the data entry (C).
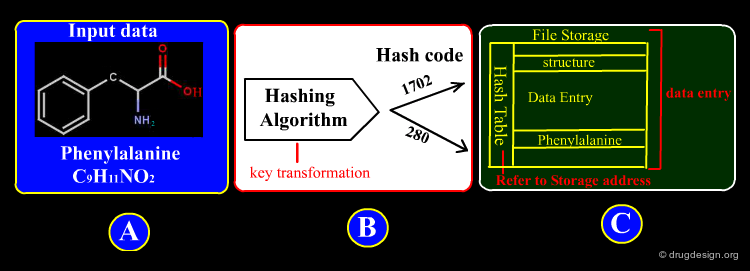
Author
Y Jane Tseng Associate Professor, Laboratory of Computational Molecular Detection and Design, Graduate Institute of Biomedical Electronics and Bioinformatics, Department of Computer Science and Information Engineering, National Taiwan University, Taiwan
articles
The Chemistry Development Kit (CDK): An Open-Source Java Library for Chemo- and Bioinformatics Steinbeck,C., Han,Y., Kuhn,S., Horlacher,O., Luttmann,E. and Willighagen,E. J. Chem. Inf. Comput. Sci. 43 2003
2D Structure Depiction Alex M. C., Paul L., and Martin S. J. Chem. Inf. Model. 46 2006
Structure Diagram Layout¶
Structure diagram layout is also known as structure diagram generation. This process generates 2D or 3D coordinates for laying out a given nomenclature. In the figure, the linear SMILES notation of 5-phenylpentanoic acid is partitioned into three substructures. Next, these substructures are converted and merged into a 2D diagram. A structure diagram is easier to understand than a text format.

Author
Y Jane Tseng Associate Professor, Laboratory of Computational Molecular Detection and Design, Graduate Institute of Biomedical Electronics and Bioinformatics, Department of Computer Science and Information Engineering, National Taiwan University, Taiwan
articles
The Chemistry Development Kit (CDK): An Open-Source Java Library for Chemo- and Bioinformatics Steinbeck,C., Han,Y., Kuhn,S., Horlacher,O., Luttmann,E. and Willighagen,E. J. Chem. Inf. Comput. Sci. 43 2003
2D Structure Depiction Alex M. C., Paul L., and Martin S. J. Chem. Inf. Model. 46 2006
3D Model Building¶
To build a 3D model of a compound, the 2D connectivity information should be given. High quality 3D coordinates can be calculated using quantum mechanical or molecular mechanics from 2D diagrams. In the example below, the 2D structure of SKI-606 was converted to a 3D model using the ACDLAB program. The general strategy used by such programs is illustrated in the following pages.

Author
Y Jane Tseng Associate Professor, Laboratory of Computational Molecular Detection and Design, Graduate Institute of Biomedical Electronics and Bioinformatics, Department of Computer Science and Information Engineering, National Taiwan University, Taiwan
Example of Strategy for 3D Model Building¶
The following is an example of algorithm for 2D->3D structure generation. Note that here, only one conformer is generated for the molecule considered.
Author
Y Jane Tseng Associate Professor, Laboratory of Computational Molecular Detection and Design, Graduate Institute of Biomedical Electronics and Bioinformatics, Department of Computer Science and Information Engineering, National Taiwan University, Taiwan
Conformer Generation¶
In the figure below is shown a typical algorithm of conformer generation. The new conformation will be compared to the set of previously generated conformers. From the comparison, the program will determine to reject or to add the conformer, into the list.

Author
Y Jane Tseng Associate Professor, Laboratory of Computational Molecular Detection and Design, Graduate Institute of Biomedical Electronics and Bioinformatics, Department of Computer Science and Information Engineering, National Taiwan University, Taiwan
General Strategy for Conformation Generation¶
There are many methods for the generation of conformers of a molecule defined in 2D. A brief overview of these methods is shown in the figure below. A multiple conformer generation procedure is not included in many programs: they produce only a single conformation of the molecule considered.

Author
Y Jane Tseng Associate Professor, Laboratory of Computational Molecular Detection and Design, Graduate Institute of Biomedical Electronics and Bioinformatics, Department of Computer Science and Information Engineering, National Taiwan University, Taiwan
Programs for Conformer Generation¶
In the following pages are illustrated some programs of conformer generation.

Author
Y Jane Tseng Associate Professor, Laboratory of Computational Molecular Detection and Design, Graduate Institute of Biomedical Electronics and Bioinformatics, Department of Computer Science and Information Engineering, National Taiwan University, Taiwan
articles
Rapid Conversion of Molecular Graphs to Three-Dimensional Representation Using the MOLGEO Program Vladimir V. Shcherbukhin and Nikolai S. Zefirov J. Chem. Inf. Comput. Sci. 33 1993
Conformational analysis using distance geometry methods Spellmeyer DC, Wong AK, Bower MJ, Blaney JM. J Mol Graph Model 15(1) 1997
DGEOM: QCPE Program QCPE-No. 590
Quantum Chemistry Program Exchange Indiana University, Bloomington IN 1997
CORINA: Automatic generation of 3D atomic coordinates for organic molecules Gasteiger J, Rudolph C, Sadowski J. Tetrahedron Computer Methodology 3 1990
book
Leach AR Reviews in Computational Chemistry VCH Publisher New York 1991
G. M. Crippen and T. F. Havel Chemometrics Series Research Studies Pr 1988
Distance Geometry (DG)¶
In Distance Geometry (DG) programs, the conformations are randomly generated using covalent distances, chirality constraints and distance ranges. The 1, 4 relationships can be determined using either torsion angles or distances (as implemented in DGEOM and MOLGEO). For example, in the structure shown below the benzene rings will be aligned, and the DG between the aliphatic atoms will be calculated.

Author
Y Jane Tseng Associate Professor, Laboratory of Computational Molecular Detection and Design, Graduate Institute of Biomedical Electronics and Bioinformatics, Department of Computer Science and Information Engineering, National Taiwan University, Taiwan
articles
Conformational analysis using distance geometry methods Spellmeyer DC, Wong AK, Bower MJ, Blaney JM. J Mol Graph Model 15(1) 1997
DGEOM: QCPE Program QCPE-No. 590
Quantum Chemistry Program Exchange Indiana University, Bloomington IN 1997
book
G. M. Crippen and T. F. Havel Chemometrics Series Research Studies Pr 1988
COBRA¶
In the COBRA program (Leach) conformational data are obtained from experimental data (e.g. X-ray crystallography) or theoretical calculations (e.g. from force-field), and are assembled in a library of predefined 3D molecular fragments. Heuristic techniques are used to automate the conformational analysis and 3D structure generation. The first step in COBRA consists of splitting the molecule into smaller fragments. For example, N,N-dimethylbenzamide is divided into different moieties.

Author
Y Jane Tseng Associate Professor, Laboratory of Computational Molecular Detection and Design, Graduate Institute of Biomedical Electronics and Bioinformatics, Department of Computer Science and Information Engineering, National Taiwan University, Taiwan
articles
COBRA Leach, A.R., Dolata, D.P., Prout, K. J. Chem. Inf. Comput. Sci. 30
CORINA¶
In CORINA (Gasteiger), molecules are separated into acyclic parts and ring systems. For small and medium-sized ring system, the geometry optimization of the ring conformations are generated from statistical and empirical data. These fragments will be reassembled and checked for steric overlaps. These ring templates are stored as lists of torsional angles. For acyclic parts, the program use data from known torsional angles stored in a torsion angle library. Below, the conformation generated for a cyclophane derivative has proven to be identical to the experimental data of this molecule.

Author
Y Jane Tseng Associate Professor, Laboratory of Computational Molecular Detection and Design, Graduate Institute of Biomedical Electronics and Bioinformatics, Department of Computer Science and Information Engineering, National Taiwan University, Taiwan
articles
CORINA: Automatic generation of 3D atomic coordinates for organic molecules Gasteiger J, Rudolph C, Sadowski J. Tetrahedron Computer Methodology 3 1990
CONCORD¶
CONCORD is a popular program for the rapid generation of 3D structures from 2D diagrams. The program is based on combination of rules and gives very low energy conformations. It was developed by the group of Perlman at the University of Texas at Austin and is commercialized by Tripos Associates (St. Louis Missouri, USA).

Author
Y Jane Tseng Associate Professor, Laboratory of Computational Molecular Detection and Design, Graduate Institute of Biomedical Electronics and Bioinformatics, Department of Computer Science and Information Engineering, National Taiwan University, Taiwan
articles
A program for the rapid generation of high quality 3D molecular structures: CONCORD (CONnections to COoRDinates) Rusinko A III, Skell JM, Balducci R, McGarity and Perlman RS St. Louis Missouri
The University of Texas at Austin and Tripos Associates
Structure Generators¶
There are three types of methods for structure generation: (1) the fragment-based (the structures are re-generated by assembling fragments using specific algorithms); (2) the rule-based and data-based (the knowledge is obtained from theorems and experimental structures); and (3) the numerical methods (the structures are built by QM (quantum mechanics), MM (molecular mechanics) and Distance Geometry calculations).
Author
Y Jane Tseng Associate Professor, Laboratory of Computational Molecular Detection and Design, Graduate Institute of Biomedical Electronics and Bioinformatics, Department of Computer Science and Information Engineering, National Taiwan University, Taiwan
Automatic Isomer Generators¶
The programs for automatic isomer generation can construct isomers that correspond to a given molecular formula and a set of fragments, with optional further restrictions (such as non-overlapping sub-structures). An example is illustrated in the figure.
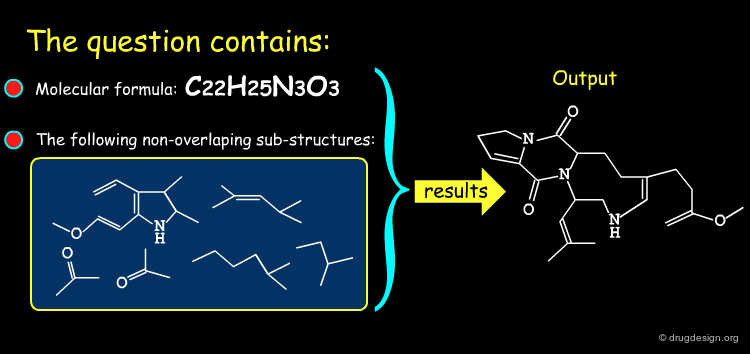
Author
Y Jane Tseng Associate Professor, Laboratory of Computational Molecular Detection and Design, Graduate Institute of Biomedical Electronics and Bioinformatics, Department of Computer Science and Information Engineering, National Taiwan University, Taiwan
MOLGEN¶
In MOLGEN, after the generation of the sketch of the constitutional isomers using a heuristic approach, the sorting of the isomers will be assessed by energy calculations subsequent to a search in the MOLGEN website. In the example below, the restrictions were: C7H8O, cycles=1 and ring size=6. A total of 387 structures was found.
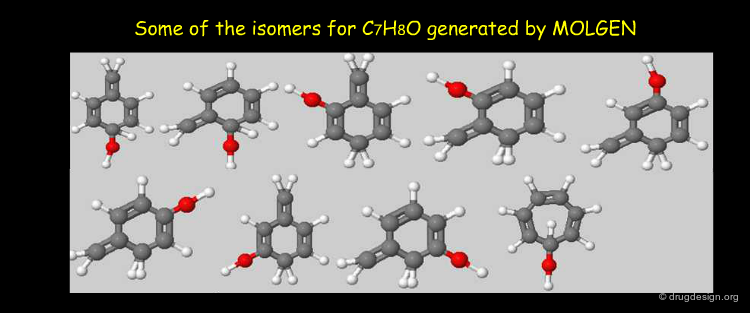
Author
Y Jane Tseng Associate Professor, Laboratory of Computational Molecular Detection and Design, Graduate Institute of Biomedical Electronics and Bioinformatics, Department of Computer Science and Information Engineering, National Taiwan University, Taiwan
articles
MOLGEN
Anal. Chim. Acta. 314 1995
Chemistry Development Kit (CDK)¶
The tools used to explore the space of isomers come from discrete mathematics such as graph theory, and combinatorics. It is based on the simulated annealing method that searches constitutional isomers with desired properties. Below are shown examples of isomers obtained with the CDK kit, for a search for C7H8O with the "benzene" restriction.

Author
Y Jane Tseng Associate Professor, Laboratory of Computational Molecular Detection and Design, Graduate Institute of Biomedical Electronics and Bioinformatics, Department of Computer Science and Information Engineering, National Taiwan University, Taiwan
Y Jane Tseng Associate Professor, Laboratory of Computational Molecular Detection and Design, Graduate Institute of Biomedical Electronics and Bioinformatics, Department of Computer Science and Information Engineering, National Taiwan University, Taiwan
articles
NMRShiftDB - Constructing a Chemical Information System with Open Source Components. Steinbeck, C.; Kuhn, S.; Krause, S. Journal of Chemical Information and Computer Sciences 43 2003
Chemical Information System (CDK) Steinbeck, C. et al J. Chem. Inf. Comput. Sci 36 1996
Names and Structures¶
A molecular structure is usually associated with many synonyms. In the example below, 2-acetyloxybenzoic acid has only one structure but is named aspirin, enterosarein etc... There are no rules for the conversion, it is therefore useful to have algorithms to do this type of conversion. Many algorithms presented before are employed and integrated into conversion algorithm programs.
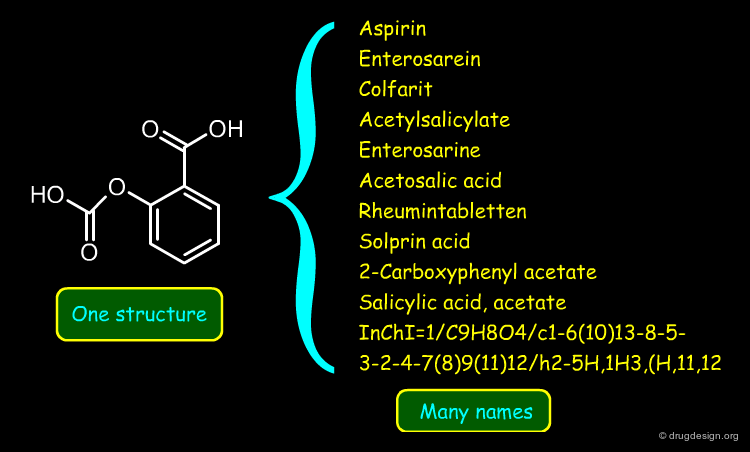
Author
Y Jane Tseng Associate Professor, Laboratory of Computational Molecular Detection and Design, Graduate Institute of Biomedical Electronics and Bioinformatics, Department of Computer Science and Information Engineering, National Taiwan University, Taiwan
book
Janusz L. W. Handbook of Chemoinformatics from Data to Knowledge WILEY-VCH Publisher 2003
Converting Structures to Names¶
A molecular structure should be represented using a name. However, naming complicated structure is a difficult task. Wisniewski proposed six phases for the algorithms of naming a molecule: (1) initialize the input structure and perceive rings (store every atom and atom vectors into a connection table and generate a hash string to record the rings); (2) recognize functional groups; (3) identify ring systems; (4) select parent structure (as root to construct a name-tree in phase 5); (5) process binary name-trees; (6) assemble chemical names from name-trees and generate resulting chemical name.

Author
Y Jane Tseng Associate Professor, Laboratory of Computational Molecular Detection and Design, Graduate Institute of Biomedical Electronics and Bioinformatics, Department of Computer Science and Information Engineering, National Taiwan University, Taiwan
articles
AUTONOM: System for Computer Translation of Structural Diagrams into IUPAC-Compatible Names. 1. General Design. J.L. Wisniewski Journal of Chemical Information and Computer Sciences 30 1990
Converting Names to Structures¶
To identify a chemical structure from a given name is difficult, even for a chemist. In general, translating names into structures can be considered as a linguistic issue. A typical algorithm is as follows: first, translate the formal or informal name to an interpretable nomenclature such as the International Union of Pure and Applied Chemistry (IUPAC) name. Second, the nomenclature is split into recognizable fragments of maximal length. Then, remove punctuation and capitalization in the fragments. Next, interpret the morphemes from each fragments, and finally, assemble these morphemes to connection tables of substructure groups to generate the structure.

Author
Y Jane Tseng Associate Professor, Laboratory of Computational Molecular Detection and Design, Graduate Institute of Biomedical Electronics and Bioinformatics, Department of Computer Science and Information Engineering, National Taiwan University, Taiwan
book
Janusz L. W. Handbook of Chemoinformatics from Data to Knowledge WILEY-VCH Publisher 2003
Available Tools for Names and Structures¶
Some software for converting structures to names or names to structures are indicated in the figure below.

Author
Y Jane Tseng Associate Professor, Laboratory of Computational Molecular Detection and Design, Graduate Institute of Biomedical Electronics and Bioinformatics, Department of Computer Science and Information Engineering, National Taiwan University, Taiwan
Copyright © 2024 drugdesign.org